From 2D Snapshots to 3D Atomic Models: A Modern Guide to Cryo-EM Virus Reconstruction for Drug Discovery
This article provides a comprehensive guide for researchers and drug development professionals on the principles, workflows, and applications of three-dimensional (3D) reconstruction of virus particles from electron microscopy (EM) images.
From 2D Snapshots to 3D Atomic Models: A Modern Guide to Cryo-EM Virus Reconstruction for Drug Discovery
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the principles, workflows, and applications of three-dimensional (3D) reconstruction of virus particles from electron microscopy (EM) images. We begin by exploring the foundational concepts of cryo-EM and single-particle analysis (SPA), establishing why this technique is transformative for structural virology. We then detail the step-by-step methodological pipeline, from sample vitrification to atomic model building, with a focus on applications in vaccine design and antiviral drug development. A dedicated troubleshooting section addresses common challenges like sample heterogeneity, preferred orientation, and resolution limits. Finally, we discuss rigorous validation metrics and comparative analyses with complementary techniques like X-ray crystallography. This guide synthesizes current best practices to empower accurate and high-resolution 3D visualization of viral structures.
The Cryo-EM Revolution: Core Principles for Visualizing Viral Architecture
Why Cryo-EM? Advantages for Studying Complex and Dynamic Virus Particles
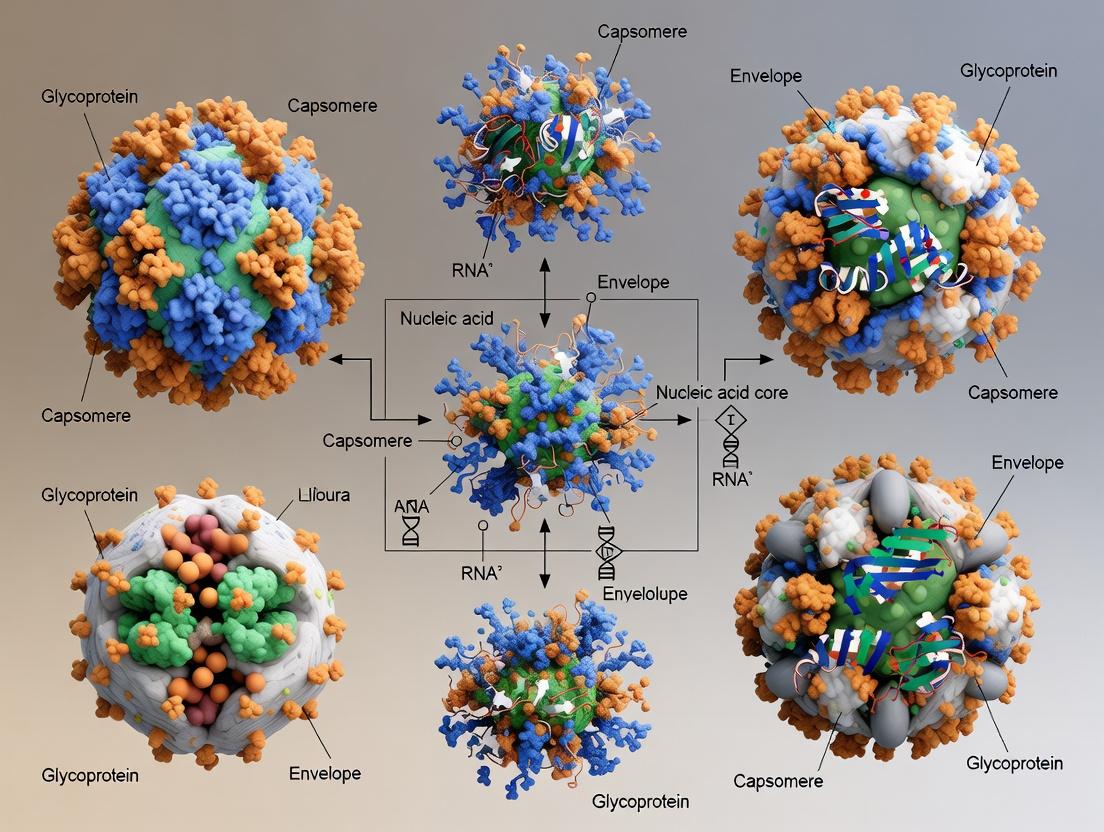
Within the broader thesis on 3D reconstruction of virus particles from electron microscopy (EM) images, Cryo-Electron Microscopy (Cryo-EM) has emerged as an indispensable technology. It enables researchers to visualize complex, pleomorphic, and dynamic viral structures in a near-native, hydrated state without the need for crystallization or heavy-metal staining. This application note details its key advantages, protocols, and resources for researchers and drug development professionals.
Key Advantages of Cryo-EM for Virology
Cryo-EM offers distinct benefits for structural virology over traditional techniques like X-ray crystallography or negative-stain EM.
Table 1: Comparative Advantages of Cryo-EM for Virus Particle Analysis
| Feature | Cryo-EM | X-ray Crystallography | Negative-Stain EM |
|---|---|---|---|
| Sample State | Hydrated, near-native vitrified ice | Crystalline, rigid | Dehydrated, stained with heavy metals |
| Size Suitability | No upper limit (viruses, complexes, cells) | Limited by crystallization | No upper limit, but sample distortion |
| Conformational Heterogeneity | Can resolve multiple states (3D Variability) | Requires homogeneous, locked conformation | Poor preservation of native conformations |
| Resolution Range | ~1.2 Å to ~10 Å (Single Particle Analysis) | Atomic (~1-3 Å) | Intermediate to low (~15-30 Å) |
| Time to Solution | Weeks to months | Often years for crystallization | Days |
| Dynamic Processes | Can capture transient states (Time-resolved Cryo-EM) | Static snapshot | Artifact-prone, not suitable |
Table 2: Quantitative Impact: Published Cryo-EM Structures of Viruses (2015-2024) Data sourced from EMDB (Electron Microscopy Data Bank) trend analysis.
| Year | Total Virus-Related EMDB Depositions | Percentage Resolved at <4Å Resolution | Notable Achievements |
|---|---|---|---|
| 2015 | ~120 | <5% | First sub-4Å structures of enveloped viruses (e.g., Dengue). |
| 2018 | ~280 | ~15% | Atomic models of giant viruses and complex capsids. |
| 2021 | ~520 | ~30% | Widespread use of Volta phase plates for small viruses. |
| 2024 | ~750 (Projected) | ~40%+ | Routine sub-3Å resolution for standard-sized viruses, enabling drug design. |
Detailed Protocols
Protocol 1: Cryo-EM Grid Preparation of Labile Enveloped Virus Particles
Objective: To vitrify purified enveloped virus samples (e.g., HIV-1, Influenza) for high-resolution Single Particle Analysis (SPA).
Materials: Purified virus suspension (≥ 0.5 mg/mL), Quantifoil or UltrAuFoil EM grids (300 mesh, R1.2/1.3 or R0.6/1), glow discharger, Vitrobot Mark IV (or equivalent), liquid ethane/propane mixture.
Procedure:
- Grid Activation: Glow discharge grids for 30-45 seconds at 15-25 mA, positive polarity, to create a hydrophilic surface.
- Sample Application: Pipette 3-4 µL of virus suspension onto the grid held by fine tweezers.
- Blotting and Vitrification: Immediately transfer grid into the Vitrobot chamber (100% humidity, 4°C or 22°C as empirically determined). Blot for 2-6 seconds with force setting -10 to +5, then plunge into liquid ethane cooled by liquid nitrogen.
- Storage: Transfer grid under liquid nitrogen to a pre-cooled storage box.
Critical Notes: Optimize blot time and humidity to achieve a thin, homogeneous ice layer without causing particle distortion or preferred orientation.
Protocol 2: High-Resolution Data Collection for Single Particle Analysis
Objective: To acquire a dataset of micrographs suitable for 3D reconstruction at high resolution.
Materials: Vitrified grid, 200-300 keV Cryo-TEM with direct electron detector (e.g., Gatan K3, Falcon 4), automated data collection software (SerialEM, EPU).
Procedure:
- Screening: Insert grid into microscope and screen for ice quality and particle density at low magnification (≤ 3,000x).
- Setup: Select suitable area. Set dose-rate to 15-25 e⁻/pixel/s on the detector. Set total exposure dose to 40-60 e⁻/Ų.
- Acquisition: Use hole or image shift-based acquisition. Collect movies in super-resolution mode (if applicable) with 30-50 frames per movie at a calibrated pixel size of 0.8-1.2 Å (e.g., 105,000x magnification).
- Defocus: Use a defocus range of -0.8 µm to -2.5 µm in staggered steps.
Protocol 3: 3D Reconstruction with Heterogeneity Analysis
Objective: To generate an initial 3D reconstruction and identify conformational states within a viral population.
Materials: Movie stack dataset, processing software (cryoSPARC, RELION).
Workflow Diagram 1: Cryo-EM Single Particle Analysis Pipeline
Diagram Title: Cryo-EM SPA & Heterogeneity Workflow
Procedure (cryoSPARC v4+):
- Preprocessing: Patch motion correction and CTF estimation on imported movies.
- Particle Picking: Use blob picker followed by 2D classification to generate templates for template-based picking.
- Initial Model: Run Ab Initio Reconstruction with 2-4 classes to generate initial maps.
- Heterogeneous Refinement: Input particles and initial models into Heterogeneous Refinement with 3-6 classes to remove junk and separate conformations.
- 3D Variability Analysis (3DVA): On a clean, homogeneous set, run 3D Variability to visualize continuous motions (e.g., capsid breathing, glycoprotein flexing).
- High-Resolution Refinement: Use Non-uniform Refinement on selected particles. Apply Local Refinement on regions of interest (e.g., spike protein).
- Model Building: Use sharpened map (B-factor -50 to -150) in Coot and Phenix for atomic model building and refinement.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Cryo-EM Virology Studies
| Item | Function & Rationale |
|---|---|
| UltrAuFoil Gold Grids (R0.6/1) | Gold foil with regular holes. Superior mechanical stability and thermal conductivity reduces ice drift vs. carbon film. |
| GraFix (Gradient Fixation) Kits | Stabilize weak protein complexes or fragile virus particles through gentle chemical cross-linking in a glycerol gradient before vitrification. |
| Fab Fragments / Nanobodies | Bind to and stabilize specific, flexible viral epitopes (e.g., envelope glycoproteins), facilitating particle alignment and high-res local reconstruction. |
| Amylose Clean Beads | For affinity purification of maltose-binding protein (MBP)-tagged viral complexes directly onto EM grids (Grid-Based Capture Method). |
| Chameleon Diamond Knives | For creating thin (50-300 nm) cryo-sections of infected cells via Cryo-Electron Tomography (Cryo-ET) to study viruses in situ. |
| 1.8 nm Undecagold-Ni-NTA Clusters | High-density fiducial markers for Cryo-ET tilt-series alignment, especially for studying virus-cell membrane interfaces. |
Diagram 2: Antibody Fragment Stabilization Strategy
Diagram Title: Fab Stabilization of Viral Glycoprotein
This document outlines the fundamental physical principles and practical protocols governing image formation in transmission electron microscopy (cryo-EM), specifically within the context of single-particle analysis (SPA) for the 3D reconstruction of virus particles. The fidelity of the final atomic model is intrinsically linked to the initial interaction between the electron beam and the specimen. Understanding electron scattering, contrast transfer, and the digitization process is therefore critical for researchers, scientists, and drug development professionals aiming to utilize cryo-EM for structure-based vaccine and antiviral drug design.
Core Physics: Scattering and Image Formation
Electron-Specimen Interaction
When a high-energy electron beam traverses a vitrified biological sample, such as a virus particle, interactions occur primarily via elastic and inelastic scattering.
- Elastic Scattering: Involves deflection without energy loss. It is coherent and provides the phase information essential for high-resolution reconstruction. It is the dominant signal for thin, frozen-hydrated specimens.
- Inelastic Scattering: Involves energy loss to the specimen, causing damage and contributing to a noisy, incoherent background. It must be minimized through low-dose imaging techniques.
The interaction is described by the specimen's electrostatic potential distribution. The scattering amplitude is given by the complex exit wave function:
ψ_exit(x,y) = exp(iσ φ_proj(x,y))
where σ is the interaction constant and φ_proj is the projected potential of the specimen.
Contrast Transfer Function (CTF)
The phase information from elastic scattering is lost at the detector. The microscope's aberrations (primarily defocus) convert these phase variations into measurable intensity variations, described by the Contrast Transfer Function (CTF). The CTF modulates the information in Fourier space (spatial frequencies).
A simplified model for the CTF is:
CTF(k) = -sin(χ(k)) * E(k)
where χ(k) = πΔλk² + 0.5πC_sλ³k⁴
and k is spatial frequency, Δ is defocus, λ is electron wavelength, and C_s is spherical aberration constant. E(k) is an envelope function accounting for temporal and spatial coherence decay.
Table 1: Key Parameters Affecting CTF in Modern Cryo-EM
| Parameter | Typical Value Range (300kV) | Effect on Image/CTF | Optimisation Goal |
|---|---|---|---|
| Accelerating Voltage | 200-300 kV | Higher voltage increases λ, reduces inelastic scattering & damage. | Maximize within stability constraints. |
| Defocus (Δ) | -0.5 to -3.0 μm | Induces contrast; oscillating CTF passes/zeros specific frequencies. | Chosen to retain first zero beyond target resolution. |
| Spherical Aberration (C_s) | ~2.7 mm (uncorrected) <0.01 mm (corrected) | Limits high-resolution transfer; causes CTF oscillations. | Minimize via aberration correctors. |
| Energy Spread (ΔE) | 0.7-1.0 eV | Dampens CTF at high frequency (temporal envelope). | Use stable FEG source, monochromator. |
| Beam Convergence | 0.05-0.1 mrad | Dampens CTF at high frequency (spatial envelope). | Use parallel (small) illumination. |
| Pixel Size (at specimen) | 0.5-1.1 Å | Must satisfy Nyquist criterion for target resolution. | At least 2x smaller than target resolution. |
Diagram Title: Image Formation Pathway in Cryo-EM
Protocols for Optimal Image Acquisition
Protocol 3.1: Low-Dose Imaging for Virus Particle Preservation
Objective: To acquire a usable image while minimizing cumulative electron dose to prevent radiation damage to the vitrified virus specimen. Materials: See "Scientist's Toolkit" (Section 5). Workflow:
- Search Mode: Navigate grid at very low magnification (~200x) and ultra-low dose (≤5 e⁻/Ų) to identify suitable ice area.
- Focus Mode: Move to adjacent hole at high magnification (~2x imaging mag), activate beam tilt for automatic defocus setting. Dose: ≤5 e⁻/Ų.
- Exposure Mode: Return to recorded coordinates. Acquire movie series (20-40 frames) with total dose of 40-60 e⁻/Ų. Use beam shutter to expose only during acquisition.
Diagram Title: Low-Dose Imaging Workflow
Protocol 3.2: CTF Estimation and Correction for SPA
Objective: To accurately determine the parameters of the CTF for each micrograph to enable subsequent phase-flip correction, essential for high-resolution 3D reconstruction. Workflow:
- Movie Processing: Align and sum movie frames (e.g., using MotionCor2) to produce a drift-corrected micrograph. Apply dose-weighting.
- Power Spectrum Calculation: Compute the 2D Fourier transform of patches of the micrograph, average to create a 1D rotational average.
- CTF Fitting: Fit the theoretical CTF curve (incorporating envelope functions) to the experimental power spectrum's Thon rings. Key fitted parameters: Defocus (Δ), Astigmatism (amplitude & angle), Amplitude Contrast ratio.
- Validation & Correction: Visually inspect fit overlaid on power spectrum. Use fitted parameters to apply phase-flip (and optionally, amplitude correction) to particle stacks in Fourier space during particle extraction or refinement.
Table 2: Quantitative CTF Fitting Quality Metrics
| Metric | Target Value | Indicates |
|---|---|---|
| Estimated Resolution Limit | <4.0 Å (for 200kV) <3.0 Å (for 300kV) | Information content at high frequency. |
| Fit Correlation Coefficient | >0.95 | Quality of match between theoretical and experimental CTF. |
| Max. Defocus Difference | <0.2 μm (within micrograph) | Ice uniformity and stability. |
| Astigmatism Amplitude | <0.1 μm | Quality of microscope alignment. |
From Analog Image to Digital Bits: Detector Technology
The conversion of electron intensity to a digital signal is critical. Direct Electron Detectors (DEDs) have revolutionized the field.
- Mechanism: Electrons hit a semiconductor pixel array, creating electron-hole pairs measured directly.
- Advantages: High Detective Quantum Efficiency (DQE ~0.8 at 300kV), fast readout enabling dose-fractionated movies, and single-electron sensitivity.
- Impact: Enables direct correction of beam-induced motion and radiation damage, pushing resolutions to near-atomic levels for virus particles.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Cryo-EM Sample & Image Preparation
| Item | Function in Virus Particle 3D Reconstruction |
|---|---|
| Quantifoil or UltrAuFoil Grids | Carbon support films with regular holes. Provide a stable, clean substrate for vitrified ice. |
| Glow Discharger | Creates a hydrophilic surface on grids to ensure even spread of sample and thin ice. |
| Vitrobot (or equivalent) | Automated plunge freezer for rapid vitrification of sample in ethane, preventing ice crystal formation. |
| Holey Gold Grids (UltrAuFoil) | Gold grids are non-magnetic and conductive, reducing drift and charging during imaging. |
| Direct Electron Detector (e.g., Gatan K3, Falcon 4) | Converts electron signal to digital with high DQE, enabling high-resolution movie acquisition. |
| 300 keV Field Emission Gun TEM | High-voltage source providing coherent electron beam with high penetration power for thick particles. |
| Spherical Aberration Corrector | Optional but increasingly common. Minimizes C_s, simplifying CTF and extending envelope function. |
| Cryo-TEM Holder | Maintains specimen at liquid nitrogen temperatures (< -170°C) during transfer and imaging. |
This article provides a conceptual and practical overview of the SPA pipeline, contextualized within a research thesis focused on the 3D reconstruction of virus particles from electron microscopy (EM) images. The pipeline enables the determination of macromolecular structures at near-atomic resolution, crucial for understanding viral mechanisms and rational drug design.
Conceptual Workflow and Key Stages
The SPA pipeline is a multi-step computational and interpretive process. The following diagram illustrates the core logical workflow from raw data to a refined 3D model.
Quantitative Metrics and Benchmarks
Successful SPA projects are guided by key quantitative benchmarks at each stage, ensuring the final reconstruction is of high quality and resolution.
Table 1: Key Quantitative Benchmarks in the SPA Pipeline
| Pipeline Stage | Key Metric | Typical Target Value (for Virus Research) | Purpose & Interpretation |
|---|---|---|---|
| Data Collection | Physical Pixel Size (Å/px) | 0.8 - 1.2 Å | Defines sampling of the specimen. Critical for achieving high resolution. |
| Defocus Range (μm) | -0.8 to -2.5 | Provides phase contrast. A range is needed for contrast transfer function (CTF) correction. | |
| Electron Dose (e⁻/Ų) | 40 - 60 | Balances signal-to-noise ratio with radiation damage. | |
| Particle Picking | Number of Initial Particles | 10^5 - 10^6 | Large datasets are required to overcome noise and conformational heterogeneity. |
| 2D Classification | Percentage of "Junk" Particles Discarded | 30-70% | Indicator of picking quality and sample purity. |
| Number of Distinct 2D Classes | 50-200 | Represents particle views and conformational states. | |
| 3D Reconstruction | Global Resolution (Å) | < 4.0 Å (for drug design) | Gold-standard FSC=0.143 criterion. Dictates level of atomic detail. |
| Local Resolution Variation (Å) | Often ± 0.5-1.5 Å of global | Flexible regions (e.g., surface glycoproteins) may be lower resolution. | |
| Map/Model Validation | Q-score (Model-to-Map Fit) | > 0.7 at core regions | Measures agreement between atomic model and cryo-EM density. |
Detailed Experimental Protocol: 3D Classification and Refinement
This protocol details a critical step for handling structural heterogeneity in virus samples, such as glycoprotein movement or genome packaging states.
Protocol: Per-particle 3D Classification and Heterogeneous Refinement in RELION/CryoSPARC
Objective: To separate a particle stack into structurally homogeneous subsets for high-resolution refinement.
Materials & Software:
- Input: A particle stack (
particles.star) and an initial 3D reference map (initial.mrc) from ab-initio reconstruction or a previous refinement. - Software: RELION-4.0 or CryoSPARC v4.0+.
- Computing: High-performance GPU cluster with > 40 GB GPU memory recommended.
Procedure:
- Initial 3D Auto-refinement: Run a single-reference 3D auto-refinement using all cleaned particles from 2D classification. Use a loose mask around the entire virus particle. This yields
refined_3d.mrcand associated particle parameters. - Generate 3D Classification References: Low-pass filter the
refined_3d.mrcto 20-30 Å resolution. Create 3-6 different copies. Optionally, apply random rotations to each to encourage divergence during classification. - Set Up 3D Classification Job:
- Number of Classes: 3 to 6. Start with fewer classes for initial exploration.
- Regularization Parameter (T): 4 to 20. Higher T allows for greater differences between classes.
- Mask: Use a broad, soft-edged mask encompassing the entire complex.
- Number of Iterations: 25-50.
- Disable Angular Searches: Set angular sampling to "fixed" or use local searches only to focus on structural differences, not orientation assignment.
- Execute and Monitor: Run the job. Monitor the log-likelihood gain to ensure convergence. Inspect intermediate class averages after 10-15 iterations.
- Analyze Results: After completion, inspect the reconstructed 3D volumes for each class.
- Selection Criteria: Discard classes showing poor features, denatured particles, or empty capsids (in the context of genome packaging).
- Homogeneity: Select the class(es) with the most homogeneous, high-resolution features (e.g., clear alpha-helical barrels, defined beta-sheets).
- Heterogeneous Refinement (CryoSPARC) / Select & Re-refine (RELION):
- In CryoSPARC, use the "Heterogeneous Refinement" job directly, which combines classification and refinement.
- In RELION, create a new particle stack (
good_particles.star) containing only particles from the selected class(es). Use this subset as input for a new, high-resolution 3D Auto-refinement job with a tighter mask and stricter convergence criteria.
- Iterate: The output from Step 6 can be used as a reference for further rounds of focused 3D classification (using a tight mask on a specific region of interest) to isolate specific conformational states.
The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for Virus SPA Sample Preparation
| Item | Function & Role in SPA Pipeline |
|---|---|
| Quantifoil or UltrAuFoil Grids | EM support grids with a regular holey carbon film. Provide a thin, stable layer of vitreous ice spanning the holes for imaging. |
| Glow Discharger (e.g., PELCO easiGlow) | Creates a hydrophilic surface on the carbon film, ensuring even sample spread and adsorption during vitrification. |
| Vitrification Device (e.g., Thermo Fisher Vitrobot Mark IV) | Rapidly plunges the EM grid into liquid ethane, freezing the hydrated sample in amorphous ice, preserving native structure. |
| Optimized Purification Buffer | A buffer (e.g., HEPES or Tris-based, 150mM NaCl) that maintains viral particle integrity, monodispersity, and stability for minutes to hours on the grid. |
| Crosslinkers (e.g., GraFix, BS3) | Optional. Gently stabilize transient complexes or flexible regions to "trap" a specific conformational state during grid preparation. |
| Fiducial Markers (e.g., 10nm Gold Beads) | Added to the sample before freezing. Provide high-contrast reference points for improved motion correction and alignment during image processing. |
| cryo-EM Screening Microscope (e.g., Talos L120C, Glacios 2) | Enables rapid assessment of grid quality, ice thickness, particle distribution, and concentration before committing to high-end data collection. |
This Application Note contextualizes key technical milestones within the broader thesis on the 3D reconstruction of virus particles from electron microscopy (EM) images. The evolution from simple 2D visualization to atomic-resolution 3D tomography has revolutionized structural virology and antiviral drug development.
Historical Milestones & Quantitative Data
The following table summarizes the quantitative progression of key parameters in virus particle EM reconstruction.
Table 1: Evolution of Resolution and Throughput in Virus EM Reconstruction
| Milestone Era (Approx.) | Key Technique | Typical Resolution (Å) | Sample Preparation Time | Data Acquisition Time | Primary Virus Study |
|---|---|---|---|---|---|
| 1959-1960s | Negative Stain EM | 20-30 Å | Minutes-Hours | Hours | TMV, Adenovirus |
| 1968-1970s | Early 3D Reconstruction (Conical Tilt) | 25-40 Å | Hours | Days | Spherical Viruses |
| 1975-1990s | Cryo-EM (Vitrification) | 10-20 Å | Hours | Days | Influenza, HIV |
| 1990s-2000s | Single Particle Analysis (SPA) | 4-8 Å | Hours | Days-Weeks | HBV, Rhinovirus |
| 2010s-Present | Direct Electron Detectors & SPA | 2-4 Å | Hours | Days | Zika, SARS-CoV-2 |
| 2015-Present | Cryo-Electron Tomography (Cryo-ET) in situ | 3-5 Å (local) | Hours | Days | Herpesviruses, HIV |
| 2018-Present | Atomic Resolution Tomography/SPA | 1.5-2.5 Å | Hours | Weeks | AAV, Rhinovirus |
Detailed Protocols
Protocol 1: Classical Negative Staining for Virus Visualization
Application: Rapid assessment of virus morphology and purity.
- Grid Preparation: Glow-discharge a 400-mesh copper grid with continuous carbon film.
- Sample Application: Apply 5-10 µL of purified virus suspension (10^11-10^12 particles/mL) to the grid for 60 seconds.
- Staining: Blot excess liquid with filter paper. Immediately apply 10 µL of 2% uranyl acetate (pH ~4.5) for 30 seconds.
- Wash & Dry: Blot stain, briefly touch grid to a droplet of distilled water, blot again, and air-dry completely.
- Imaging: Insert grid into TEM. Image at 80-100 kV under low-dose conditions (~20 e/Ų).
Protocol 2: High-Resolution Single Particle Analysis (SPA) Cryo-EM
Application: Determining near-atomic resolution 3D structures of purified, homogeneous virus particles.
- Vitrification: Use a vitrification robot. Apply 3 µL of purified virus sample to a freshly glow-discharged Quantifoil R1.2/1.3 Au grid. Blot for 3-4 seconds at 100% humidity, 4°C, and plunge-freeze into liquid ethane.
- Screening: Assess grid quality (ice thickness, particle distribution) using a 200 kV screening cryo-TEM.
- High-Resolution Data Collection: On a 300 kV cryo-TEM equipped with a Gatan K3 direct electron detector:
- Set nominal magnification to 105,000x (pixel size 0.826 Å).
- Use a defocus range of -0.8 to -2.5 µm.
- Collect ~40 frames per exposure with a total dose of 50 e/Ų fractionated across frames.
- Acquire 5,000-10,000 micrographs in automated session (~48-72 hours).
- Image Processing (Workflow): Follow the logical pipeline below.
Diagram Title: Cryo-EM SPA Workflow for Virus Reconstruction
Protocol 3: Subtomogram Averaging for In-Situ Virus Capsids
Application: Determining high-resolution structure of virus particles within cellular context.
- Sample Preparation: Infect cells (e.g., HEPA) on gold EM carriers. High-pressure freeze at peak infection. Prepare ~200 nm lamellae using cryo-FIB-SEM.
- Tilt-Series Acquisition: Acquire tilt-series from -60° to +60° with 2° increment at 300 kV. Use dose-symmetric scheme, total dose <120 e/Ų.
- Tomogram Reconstruction: Align tilt-series using gold fiducials. Reconstruct tomogram via weighted back-projection (e.g., IMOD).
- Subtomogram Averaging: Manually or template-pick particle sub-volumes from multiple tomograms. Align, classify, and average using Relion or M.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for High-Resolution Virus Cryo-EM
| Item | Function & Rationale |
|---|---|
| Quantifoil R1.2/1.3 Au Grids | Holey carbon film on gold mesh. Gold provides better thermal conductivity than copper, reducing beam-induced motion. |
| Uranyl Formate / Uranyl Acetate | High-contrast negative stain for rapid validation of sample quality and particle presence. |
| Liquid Ethane | Cryogen for rapid vitrification of aqueous samples, preventing crystalline ice formation. |
| C-flat or UltrAuFoil Grids | Alternative grids with reproducible holey patterns or gold foil, optimizing ice uniformity. |
| GraFix (Gradient Fixation) | Reagents for stabilizing weak protein complexes via glycerol gradient and gentle crosslinking prior to EM. |
| Ammonium Molybdate | Negative stain with near-neutral pH, preserving fragile structures better than uranyl acetate. |
| Fiducial Gold Beads (10-15 nm) | For precise alignment of tilt-series in cryo-ET. Provide high-contrast markers. |
| Tris(2-carboxyethyl)phosphine (TCEP) | Reducing agent added to purified virus samples to prevent aggregation during grid preparation. |
| ChamQ SYBR Colorimetric PCR Mix | Used for quantitative PCR (qPCR) to precisely titer virus stock concentrations before grid freezing. |
This application note is framed within a broader thesis on the 3D reconstruction of virus particles from electron microscopy (EM) images. The high-resolution structural determination of viruses is pivotal for understanding viral life cycles, host-pathogen interactions, and for rational drug and vaccine design. The single-particle cryo-EM workflow has revolutionized this field, with several software packages forming the essential computational ecosystem for processing the complex image data into atomic models.
Core Software Ecosystem: Quantitative Comparison
The following table summarizes key quantitative metrics and characteristics of the leading software packages used for high-resolution virus structure determination.
Table 1: Core Software for Single-Particle Cryo-EM 3D Reconstruction
| Software | Current Version (as of 2024) | Primary Licensing Model | Key Algorithmic Strength | Typical Benchmark Resolution (Virus Studies) | Execution Environment |
|---|---|---|---|---|---|
| RELION | 4.0 | Open-source (GPL) | Bayesian particle polishing, CTF refinement, E2E deep learning in 5.0 beta | ~1.8 – 3.0 Å | Standalone (CPU/GPU), Scipion |
| cryoSPARC | v4.4 | Commercial (subscription), academic tier | Ab-initio reconstruction, heterogeneous refinement, live processing | ~1.9 – 3.2 Å | Cloud/Server (GPU-centric) |
| EMAN2/SPARX | 2.9 | Open-source (GPL) | Extensive toolset for initial processing, 2D classification, helical | ~2.5 – 4.0 Å | Standalone |
| cisTEM | 1.0.0-beta | Open-source (GPL) | User-friendly, integrated workflow from movies to maps | ~2.2 – 3.5 Å | Standalone (CPU/GPU) |
| SPHIRE-crYOLO | 1.8.x | Open-source (GPL) | Deep learning-based particle picking (integrated with RELION/SPARX) | N/A (Picking Tool) | Standalone/Plugin |
Application Notes & Detailed Protocols
Protocol: High-Resolution Reconstruction of an Icosahedral Virus using RELION 4.0
Objective: To obtain a near-atomic resolution 3D reconstruction from cryo-EM micrographs of a purified icosahedral virus sample.
Reagents & Materials:
- Purified virus suspension (>0.5 mg/mL) in appropriate buffer.
- Quantifoil R1.2/1.3 or UltrAuFoil 300-mesh gold grids.
- Vitrobot Mark IV (or equivalent) for plunge-freezing.
- Data: 5,000-10,000 dose-fractionated micrographs collected at 300 keV with a K3 or Falcon4 direct electron detector in counting mode, at a nominal magnification of 81,000x (~0.55 Å/pixel), with a total dose of 40-50 e⁻/Ų.
Procedure:
Pre-processing:
- Use
relion_run_motioncorrfor beam-induced motion correction andrelion_run_ctffindfor CTF estimation on the dose-weighted micrographs. - Manually inspect micrographs for ice quality, CTF fit, and astigmatism. Discard poor-quality images.
- Use
Initial Model Generation (Ab-initio):
- Perform reference-free particle picking using
relion_autopickwith Laplacian-of-Gaussian (LoG) filter. Extract ~500,000 particles with a large box size. - Conduct several rounds of 2D classification in RELION to remove non-particle picks and contaminants. Select clean class averages.
- Use stochastic gradient descent (SGD) in RELION (
relion_refine) with de novo initial model generation from a random blob, imposing icosahedral (I1) symmetry.
- Perform reference-free particle picking using
High-Resolution 3D Refinement:
- Refine the selected particles against the initial model using Bayesian polishing and per-particle CTF refinement options enabled.
- Run a final 3D auto-refine with a tight mask and imposed symmetry.
- Generate a post-processed map using
relion_postprocesswith automatic B-factor sharpening.
Validation:
- Use the
relion_image_handler --bfactorcommand to estimate the global B-factor. - Calculate the gold-standard Fourier Shell Correlation (FSC) between two independently refined half-maps. Report the FSC=0.143 resolution.
- Use the
Diagram 1: RELION 4.0 Icosahedral Virus Workflow
Protocol: Heterogeneous Analysis of Viral Conformations using cryoSPARC
Objective: To disentangle multiple conformational or compositional states of a complex virus particle (e.g., genome-packed vs. empty capsids).
Procedure:
Import and Pre-process:
- Import motion-corrected micrographs (e.g., from MotionCor2) and CTF parameters (e.g., from CTFFIND4) into a cryoSPARC project.
- Use the Patch CTF job for local refinement of CTF parameters.
Heterogeneous Refinement:
- Extract particles using a template picker or from RELION. Import them into cryoSPARC.
- Perform an Ab-initio Reconstruction job with 3 output classes and no symmetry (C1) to generate starting models for different states.
- Run a Heterogeneous Refinement job using the ab-initio models as inputs, along with the particle set. This will iteratively sort particles into the distinct classes.
High-Resolution Reconstruction of States:
- Extract the particle stack for each well-defined class from the heterogeneous refinement.
- Perform a Non-uniform Refinement (with or without imposed symmetry) on each homogeneous subset to achieve high-resolution maps for each conformational state.
- Use the Local Resolution and FSC tools for validation.
Diagram 2: cryoSPARC Heterogeneous Analysis Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Cryo-EM of Virus Particles
| Item | Function & Rationale |
|---|---|
| UltrAuFoil Gold Grids (300 mesh) | Gold supports improve thermal conductivity and stability during imaging, reducing beam-induced motion. The holes are pre-fabricated, yielding more reproducible ice thickness. |
| 1.2/1.3 µm Holey Carbon Films (Quantifoil) | The standard for high-resolution work. The thin, continuous carbon across holes can provide support for fragile particles. |
| Ammonium Molybdate (2%) | A common negative stain for quick initial validation of virus sample purity, concentration, and structural integrity by room-temperature EM. |
| n-Dodecyl-β-D-Maltoside (DDM, 0.01%) | A mild detergent used during virus purification or grid preparation to disrupt lipid vesicles or aggregates without disassembling the viral capsid. |
| Tris(2-carboxyethyl)phosphine (TCEP) | A reducing agent added to purification buffers to prevent disulfide-mediated aggregation of viral surface proteins, promoting monodispersity. |
| Glycerol (5-10%) | May be included in the final buffer before grid freezing for some enveloped viruses to act as a cryo-protectant, improving vitreous ice quality. |
| Aurion Biotinylated Nanogold (5 nm) | Fiducial markers for tomography or for localizing specific components on virus surfaces when conjugated to streptavidin-labeled antibodies. |
Step-by-Step Reconstruction Workflow: From Sample Prep to Atomic Model
Within the broader thesis on the 3D reconstruction of virus particles from EM images, achieving high-resolution single-particle analysis (SPA) is fundamentally constrained by the quality of the initial specimen. Sample preparation is the critical, non-negotiable first step. Mastery of vitrification, grid selection, and particle density optimization directly dictates the success of downstream data collection and reconstruction, impacting studies of viral structure, function, and drug binding for development professionals.
Protocol 1: Optimized Vitrification for Intact Virus Particles
This protocol details the plunge-freezing of purified virus suspensions to preserve native-state structure in a thin layer of vitreous ice.
Materials:
- Purified virus suspension (≥ 0.5 mg/mL, in preferred buffer).
- Quantifoil or C-flat EM grids (300 mesh, Au, R 1.2/1.3 or 2/1).
- Glow discharger (e.g., PELCO easiGlow).
- Plunge freezer (e.g., Thermo Fisher Vitrobot Mark IV, Leica EM GP).
- Filter paper (standard grade, blotting).
- Ethane gas and liquid nitrogen.
- Cryo storage boxes and dewar.
Procedure:
- Grid Preparation: Glow discharge grids for 30-60 seconds at 15-25 mA, negative polarity, to render the carbon support hydrophilic.
- Freezer Setup: Pre-condition the Vitrobot chamber to 100% humidity and a temperature of 4-10°C (to minimize evaporation). Fill the ethane container to create a liquid ethane slurry.
- Sample Application: Pipette 3-5 µL of virus suspension onto the glow-discharged side of the grid mounted in the tweezers.
- Blotting: Initiate the automated blot cycle. Typical parameters: Blot time 3-5 seconds, Blot force 0-5, Wait time 0 seconds. Critical: Optimize blot time to achieve an ice thickness slightly greater than the virus diameter.
- Plunge & Vitrification: After blotting, immediately plunge the grid at maximum speed into liquid ethane. Hold for a few seconds, then transfer under liquid nitrogen to a pre-cooled storage box.
- Storage: Keep grids immersed in liquid nitrogen until loading into the microscope.
Protocol 2: Systematic Screening for Optimal Particle Density
This method provides a quantitative framework for adjusting sample concentration and preparation parameters to achieve ideal particle distribution for automated data collection.
Materials:
- Prepared cryo-EM grids from Protocol 1.
- Cryo-TEM equipped with a direct electron detector.
- Data collection software (e.g., SerialEM, EPU).
Procedure:
- Initial Screening: Load a grid and at low magnification (e.g., 100x), identify grid squares with suitable, thin ice.
- Micrograph Acquisition: Acquire 10-20 random micrographs at your target SPA magnification (e.g., 81,000x, corresponding to ~1.0 Å/pixel) with a minimal dose (~1 e⁻/Ų).
- Particle Counting: Using a quick manual pick or automated picking tool in real-time, count the number of intact, well-separated virus particles per micrograph.
- Density Calculation & Adjustment: Calculate particles per square micron. Compare to the target range (see Table 1). Adjust the original sample concentration or blot conditions accordingly and prepare a new grid.
- Iterate: Repeat screening until the majority of grid squares yield the target particle density.
Data Presentation
Table 1: Quantitative Parameters for Optimal Single-Particle Analysis of Virus Particles
| Parameter | Optimal Range | Impact on Reconstruction | Notes for Virus Studies |
|---|---|---|---|
| Ice Thickness | 1.2 - 1.5 x particle diameter | Thick ice increases noise, thin ice denatures particles. | For a 100nm virus, target 120-150nm ice. |
| Particle Density | 80 - 150 particles/µm² | Too low: inefficient collection. Too high: particle overlap, mis-picking. | Enveloped viruses may require lower density. |
| Sample Concentration | 0.5 - 3.0 mg/mL (empirical) | Directly influences particle density in ice. | Buffer composition drastically affects adsorption. |
| Defocus Range | -0.8 µm to -2.5 µm (staggered) | Provides necessary contrast & CTF information. | Use closer focus (-0.8 to -1.5 µm) for small viruses. |
| Blot Time (Vitrobot) | 2 - 6 seconds | Primary control for ice thickness. | Humidity and temperature are critical variables. |
Table 2: Cryo-EM Grid Selection Guide for Virology
| Grid Type | Hole Size/Pattern | Key Advantages | Best For |
|---|---|---|---|
| Quantifoil R 1.2/1.3 | 1.2 µm holes, 1.3 µm spacing | Proven reliability, extensive literature. | General virus SPA, standard workflows. |
| C-flat CF-2/1 | 2 µm holes, 1 µm spacing | Larger ice area, good support. | Larger viruses (>150 nm) or asymmetric complexes. |
| UltrAuFoil R 0.6/1 | 0.6 µm holes, 1 µm spacing | Gold foil, superior conductivity, reduced drift. | High-resolution studies of small/rigid viruses. |
| Lacey Carbon | Irregular holes | Very large, continuous ice areas. | Initial screening, filamentous viruses. |
Visualizations
Diagram 1: Cryo-EM Sample Prep & Screening Workflow
Diagram 2: Particle Density & Ice Quality Optimization Logic
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents & Materials for Viral Cryo-EM Sample Prep
| Item | Function in Protocol | Critical Consideration for Virology |
|---|---|---|
| EM Grids (Quantifoil/C-flat, Au) | Physical support for the vitreous ice film. | Gold grids minimize magnetic interactions. Hole size must accommodate virus diameter. |
| Glow Discharger | Creates hydrophilic carbon surface for even sample spread. | Over-discharging can aggregate viruses. Optimize time/polarity for your virus. |
| Plunge Freezer (Vitrobot) | Provides controlled humidity, temperature, and blotting for reproducible vitrification. | 4-10°C chamber temp helps preserve lipid envelopes. |
| Liquid Ethane | Cryogen with high heat capacity for rapid vitrification. | Must be maintained just above melting point for slurry formation. |
| Purified Virus Buffer | Stabilizes native virus structure during grid preparation. | Must be compatible with vitrification. Avoid high salt (>300 mM) or glycerol. |
| Cryo Storage Boxes | Secure, labeled long-term storage under liquid nitrogen. | Pre-cool in LN2 vapor to prevent thermal shock and ice contamination. |
Within the broader thesis on the 3D reconstruction of virus particles from electron microscopy (EM) images, the data acquisition strategy is the foundational determinant of resolution. This document details application notes and protocols for implementing automated data collection, dose fractionation, and defocus management to optimize high-resolution single-particle analysis (SPA) for structural virology and drug discovery.
Core Strategies & Quantitative Comparison
The interplay between automation, dose fractionation, and defocus management dictates data quality and throughput. The following table summarizes the key parameters and their quantitative impact on 3D reconstruction.
Table 1: Quantitative Impact of Acquisition Strategies on Virus Particle Reconstruction
| Strategy Parameter | Typical Value/Range | Primary Impact on Reconstruction | Quantitative Goal for Viruses (∼200-1000 Å) |
|---|---|---|---|
| Total Electron Dose | 40-80 e⁻/Ų | High dose increases SNR but causes radiation damage. | ≤ 60 e⁻/Ų for enveloped viruses; ≤ 80 e⁻/Ų for capsids. |
| Dose Fractionation (Frames) | 40-60 frames | Enables dose weighting & motion correction. | 40-50 frames for optimal balance of correction vs. file size. |
| Defocus Range (Target) | -0.8 µm to -2.5 µm | Provides contrast & phase information transfer. | -1.2 µm to -2.2 µm (staggered). Minimum 3-5 defocus groups. |
| Pixel Size at Specimen | 0.8 - 1.2 Å | Determines Nyquist limit. | ≤ 1.1 Å for sub-3Å target resolution. |
| Autoloader Capacity | 12 grids | Enables unsupervised, multi-grid collection. | 24-48h of continuous acquisition. |
| Particles per Micrograph | 50-200 particles | Impacts data set size and speed. | >100 particles/image improves throughput. |
| Estimated Data for 3.5Å Map | 5,000-10,000 particles | Final dataset size depends on particle size & symmetry. | 3,000-5,000 particles for an icosahedral virus (≥60 symmetry). |
Defocus Scheme Optimization
A pre-calibrated defocus scheme is critical. The following table provides a protocol for a standard multi-grid defocus strategy.
Table 2: Staggered Defocus Protocol for Multi-Grid Acquisition
| Grid Position | Target Defocus 1 (µm) | Target Defocus 2 (µm) | Target Defocus 3 (µm) | Purpose |
|---|---|---|---|---|
| Hole 1, A1 | -1.0 | -1.5 | -2.0 | Calibration & CTF assessment. |
| Primary Data Collection | -1.2 | -1.8 | -2.4 | Main scheme for high-resolution information transfer. |
| High-Contrast (Large Virus) | -1.5 | -2.5 | -3.5* | For very large or low-contrast specimens (e.g., enveloped virions). |
| Near-Focus (High-Res) | -0.8 | -1.0 | -1.2 | Target for sub-2Å reconstructions (requires very stable specimens). |
Note: Use -3.5 µm defocus sparingly as it attenuates high-resolution signals.
Detailed Experimental Protocols
Protocol: Automated Multi-Grid Data Acquisition with Dose Fractionation
Objective: To acquire a large, high-quality dataset of virus particles over 24-48 hours with minimal intervention. Materials: Vitrified specimen grids (up to 12), 200-300 keV cryo-TEM with autoloader, phase plate (optional), automated acquisition software (e.g., SerialEM, EPU, Leginon).
Procedure:
- System Calibration:
- Align microscope for parallel illumination. Calimate beam tilt for coma-free alignment.
- Calibrate image shift, beam tilt, and defocus for the preset magnification (e.g., 81,000x for 1.06 Å/pixel).
- Set up dose-symmetric or dose-optimal scheme for frame-based acquisition (e.g., 40 frames, 1.25 e⁻/Ų/frame).
Grid Loading & Screening:
- Load up to 12 grids into the autoloader cassette under liquid nitrogen.
- Initiate automated loading and screening. For each grid:
- Collect a low-magnification (e.g., 280x) atlas map.
- Identify squares/holes with optimal ice thickness (50-80 nm) and high particle density.
- Register these positions for later data collection.
Acquisition Template Setup:
- Define a multi-hole acquisition pattern per square. Use beam-image shift to visit multiple holes per stage movement.
- Set the staggered defocus scheme (per Table 2). Apply an offset (e.g., ±0.2 µm) to each target to increase defocus diversity.
- Enable focus-and-tracking: acquire a preview image, calculate defocus, apply correction, track specimen shift, then expose.
- Set total dose to 60 e⁻/Ų (fractionated across frames). Use a small (∼1 µm) beam for parallel illumination.
Unsupervised Collection & Monitoring:
- Initiate the run. The software will cycle through grids, squares, and holes.
- Monitor progress remotely. Software should flag and skip holes with poor ice, contamination, or drift.
Data Transfer & Backup:
- Configure automatic transfer of dose-fractionated movies (e.g., in MRC or TIF format) to a high-speed storage server.
- Implement a live backup system.
Protocol: Defocus Management and CTF Assessment
Objective: To ensure accurate defocus targeting and immediate assessment of CTF parameters for quality control. Materials: Acquired micrographs (or movie frames), CTF estimation software (e.g., CTFFIND4, Gctf, patchCTF).
Procedure:
- On-the-Fly CTF Estimation:
- Configure acquisition software to estimate CTF from the first or motion-corrected average of each micrograph immediately after exposure.
- Key output parameters: Defocus (µm), Astigmatism (Å), Cross-correlation score, Estimated maximum resolution.
Quality Control Filtering:
- Set acceptance criteria: |Astigmatism| < 500 Å; Cross-correlation score > 0.8 at 5 Å; Ice thickness from fit.
- Micrographs failing criteria are flagged. The software can adjust targeting to avoid poor areas.
Defocus Group Assignment for Processing:
- Post-collection, bin micrographs into defocus groups (e.g., -1.2 ± 0.1 µm, -1.8 ± 0.1 µm, -2.4 ± 0.1 µm).
- This grouping is used during particle extraction and refinement to account for CTF variation.
Visualizations
Diagram 1: Automated cryo-EM data acquisition workflow.
Diagram 2: Dose fractionation and movie processing pathway.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for High-Resolution Virus cryo-EM
| Item | Function/Description | Example Product/Note |
|---|---|---|
| Quantifoil R 1.2/1.3 or UltrAuFoil Holey Grids | Support film with regular holes for vitrification. UltrAuFoil (gold) offers better thermal conductivity and stability. | Quantifoil R 1.2/1.3 300 mesh; UltrAuFoil R 1.2/1.3 300 mesh. |
| Plasma Cleaner (GentleGlow or equivalent) | Hydrophilizes grid surface immediately before blotting to ensure even ice distribution. | Settings: 5-15 mA, 30-60 seconds, air/oxygen mix. |
| Vitrobot Mark IV or GP2 Plunger | Automated, environmentally controlled instrument for reproducible vitrification. | Set to 100% humidity, 4-10°C, optimized blot force/time. |
| C-Flat or C-Multi Grid Boxes | Secure, magnetic grid storage cassettes compatible with most autoloaders. | Prevents grid damage and mixing during transfer under LN₂. |
| Liquid Nitrogen Dewar with Autoloader Cassette | Long-term storage and automated transfer of grids into the microscope. | Ensures grids remain below -175°C at all times. |
| Focused Ion Beam (FIB) Mill (optional) | For creating lamellae of virus-infected or complex cellular samples (cryo-electron tomography). | Essential for in-situ structural virology studies. |
| Gatan K3 or Falcon 4 Direct Electron Detector | High-speed, low-noise camera enabling dose fractionation. | Operated in counting or super-resolution mode. |
| Phase Plate (Volta or Zach) | Increases contrast at low defocus, allowing data collection closer to focus. | Particularly useful for small viruses (<300 Å). |
| SerialEM, EPU, or Leginon Software | Automated data acquisition software packages. | SerialEM is open-source; EPU is commercial (Thermo Fisher). |
| CryoSPARC Live, Warp, or RELION-4 | Software for on-the-fly processing, motion correction, CTF estimation, and 2D classification. | Enables real-time feedback on data quality during acquisition. |
Within the broader thesis on the 3D reconstruction of virus particles from electron microscopy (EM) images, the stages of particle picking, 2D classification, and initial model generation form the critical computational foundation. This pipeline transforms raw, noisy micrographs into structured, interpretable data suitable for high-resolution 3D reconstruction. The fidelity of the final atomic model, essential for understanding viral pathogenesis and rational drug design, is directly contingent upon the rigor applied in these initial image processing steps.
Particle Picking: Automated Detection of Viral Particles
Particle picking isolates individual virus particles from cryo-EM micrographs. Modern approaches predominantly use deep learning due to superior accuracy over template-based methods.
Current Quantitative Performance Metrics (2024-2025)
The following table summarizes the performance of widely used particle picking tools as reported in recent literature and benchmarks.
Table 1: Performance Comparison of Major Particle Picking Algorithms
| Software/Tool | Core Algorithm | Reported Precision | Reported Recall | Key Advantage | Best Suited For |
|---|---|---|---|---|---|
| cryoSPARCBlob Picker / Template Picker | Laplacian-of-Gaussian / NCC | ~70-85% | ~75-90% | Fast, integrated workflow | Initial sweeps, homogeneous samples |
RELION-4relion_autopick |
Deep learning (CryoLOKO) | ~90-95% | ~90-95% | High accuracy, low false positive | Challenging datasets, heterogeneous backgrounds |
Topaztopaz pick |
Deep learning (ConvNet) | 85-93% | 88-94% | Trains on small labeled data | Datasets with unique features or contaminants |
Warpwarp-picker |
Deep learning (TileNet) | 88-94% | 85-92% | Fully automated, on-the-fly | Large-scale, streaming processing |
EMAN2.91e2boxer.py |
Neural Network (DeepPicker) | 80-90% | 82-90% | Strong GUI integration | Manual curation & interactive use |
NCC: Normalized Cross-Correlation. Precision: Correct picks / Total picks. Recall: Correct picks / Total true particles.
Detailed Protocol: Deep Learning-Based Picking with RELION-4
Objective: To generate a high-fidelity set of particle coordinates from cryo-EM micrographs with minimal false positives and false negatives.
Materials & Software:
- RELION-4.0 or later.
- A set of motion-corrected and dose-weighted micrographs (
.mrcformat). - Approximately 100-500 manually picked particles for training.
Procedure:
Manual Seed Generation:
- Launch the RELION GUI and use the
Manual pickingjob on 5-10 representative micrographs. - Carefully pick ~200-500 clearly identifiable particles. Avoid ice contamination and carbon edges. Save as a STAR file (
manual_picks.star).
- Launch the RELION GUI and use the
Training the Deep Learning Model:
- Run a
Particle pickingjob, selectDeep learning (CryoLOKO). - Input the micrographs and the
manual_picks.staras training data. - Set parameters: Box size (e.g., 1.5x particle diameter), minimum inter-particle distance (e.g., 80% of diameter), and a conservative initial threshold (e.g., 0.1).
- Execute. The model will train for several hundred iterations, monitoring loss.
- Run a
Automated Picking and Curation:
- Once training converges, apply the trained model to the full dataset.
- Adjust the picking threshold (e.g., 0.3-0.5) to balance yield and purity based on the preview.
- Extract particles using the generated coordinates. Perform an initial 2D classification (see Section 3) on a random subset (e.g., 50,000 particles).
- Use clean 2D class averages as
Referencesfor a subsequentTemplate-based pickjob to capture particles missed by the deep learner.
Visualization: Particle Picking Workflow
Title: Deep Learning-Enhanced Particle Picking Protocol
2D Classification: Sorting and Cleaning
2D classification aligns extracted particles and groups them into visually similar classes, removing junk particles (ice, detergent, broken particles).
Quantitative Outcomes and Metrics
Table 2: Expected Outcomes from 2D Classification for Virus Samples
| Parameter | Typical Range / Value | Interpretation & Goal |
|---|---|---|
| Number of Classes | 50 - 200 | Enough to separate orientations/defects, not so many that classes are noisy. |
| Particles per Class | 100 - 5000 (varies) | Well-populated classes yield high-SNR averages. |
| Final Yield | 60% - 90% of raw picks | Depends on sample purity and picking accuracy. Target >70% for good prep. |
| Class Variance | Monitor in GUI | High intra-class variance suggests misalignment or heterogeneity. |
| Key Visual Signatures | - | Good: Consistent capsid features, clear icosahedral edges. Bad: Fuzzy rings, featureless blobs, straight lines (ice). |
Detailed Protocol: Iterative 2D Classification in cryoSPARC
Objective: To generate clean, high-signal-to-noise 2D class averages and select a subset of particles for 3D reconstruction.
Materials & Software:
- cryoSPARC v4.2+.
- Extracted particle stack (e.g., from Section 2).
Procedure:
Initial Classification (Broad Separation):
- Create a
2D Classificationjob. Input the particle stack. - Use a large number of classes (e.g., 100-150), moderate batch size, and medium iterations (e.g., 40-50).
- Set
Class similarityto high. This performs a first-pass separation of obvious junk, intact particles, and distinct orientations.
- Create a
Manual Curation & Selection:
- Visually inspect all class averages. Select classes showing clear, high-resolution features of the virus capsid.
- Select good classes and use the
Select from 2D Classesjob to create a new particle stack.
Refined Classification (Focus on Quality):
- Run a new
2D Classificationjob on the cleaned stack. - Use a smaller number of classes (e.g., 50-80) and increase iterations (e.g., 60-80) for finer separation.
- This round may separate different particle orientations or conformational states.
- Run a new
Final Selection for Heterogeneous Reconstruction (if needed):
- If the virus exhibits asymmetry (e.g., portal complex, genome packaging), carefully group classes by these features.
- Create multiple particle subsets representing different states for downstream 3D variability analysis or separate reconstructions.
Visualization: Iterative 2D Classification Workflow
Title: Iterative 2D Classification for Particle Curation
Initial Model Generation: From 2D to 3D
Initial model creation is a critical step that determines the success of high-resolution refinement. It must be free of bias and at a sufficient resolution to align particles correctly.
Quantitative Guidelines for Initial Models
Table 3: Initial Model Generation Methods and Applications
| Method | Typical Resolution Range | Minimum Particle # | Key Strength | Major Risk / Limitation |
|---|---|---|---|---|
| Stochastic Gradient Descent (cryoSPARC) | 20-30 Å | ~5,000 | Robust, ab-initio, no template. | May fail on very symmetric/small particles. |
| Random Phase / 3D Initial Model (RELION) | 25-40 Å | ~10,000 | Truly unbiased start. | Can converge to incorrect model (bias). |
| Common Lines (EMAN2 e2initialmodel) | 25-35 Å | ~3,000 | Works with very few particles. | Sensitive to inaccurate CTF parameters. |
| Homologous Model (Low-pass filtered) | N/A (Template) | N/A | Fast, reliable if template correct. | Introduction of reference bias. |
| Symmetry Expansion & Averaging | <20 Å (post-refinement) | Depends on symmetry | Boosts signal for small/asymmetric features. | Computationally intensive. |
Detailed Protocol: Ab-Initio Reconstruction in cryoSPARC
Objective: To generate a de novo, unbiased 3D initial model from a cleaned particle set.
Materials & Software:
- cryoSPARC v4.2+.
- A cleaned particle stack from 2D classification.
- Corresponding CTF parameters.
Procedure:
Job Setup:
- Create an
Ab-Initio Reconstructionjob. Input the particle stack and CTF parameters. - Set the number of output volumes to
3. Running multiple models allows diagnosis of stability.
- Create an
Parameter Configuration:
- Symmetry: Apply the correct point-group symmetry if known (e.g., I1 for asymmetric, I3 for 3-fold, etc.). For a novel virus, start with C1.
- Batch Size: Use the default (~4000). Reduce if memory is limited.
- Number of Particles per Model: Use all available particles for the best signal.
- Resolution Limits: Set initial low-pass filter to 15-20 Å to prevent overfitting.
Execution and Validation:
- Run the job. Monitor the per-iteration resolution estimates and Fourier Shell Correlation (FSC) between the independently reconstructed models.
- Success Criteria: At least 2 of the 3 models converge to a similar structure with recognizable capsid features. Their FSC should indicate a resolution better than 30-35 Å.
- Failure Mode: All models are featureless spheres or differ drastically. This indicates persistent junk particles, severe misalignment in 2D, or insufficient particle number.
Selection and Next Steps:
- Select the best model (most detailed, highest resolution FSC) as the reference for
Heterogeneous Refinementto perform a final clean-up, or for directHomogeneous Refinement.
- Select the best model (most detailed, highest resolution FSC) as the reference for
Visualization: Initial Model Generation and Validation
Title: Ab-Initio 3D Model Generation and Quality Control
The Scientist's Toolkit: Essential Research Reagents & Software
Table 4: Key Reagent Solutions and Computational Tools for Cryo-EM Image Processing Core
| Item / Software | Category | Function & Purpose in Protocol |
|---|---|---|
| cryoSPARC (Live) | Software Suite | Integrated platform for processing from micrographs to 3D refinement. Used for 2D classification and Ab-Initio reconstruction. |
| RELION-4 | Software Suite | Bayesian approach for high-resolution refinement. Its deep learning picker (CryoLOKO) is state-of-the-art for particle picking. |
| CTFFIND4 / Gctf | Software Tool | Determines the Contrast Transfer Function (CTF) parameters of each micrograph, essential for correction. |
| PyEM / UCSF pyem | Python Toolkit | Suite of scripts (e.g., cryodrgn) for advanced processing, including deep learning analysis and heterogeneity. |
| Topaz | Software Tool | Deep learning-based particle picking, especially useful when retraining on specific datasets. |
| Blik / ChimeraX | Visualization | Interactive 3D visualization and analysis of maps, models, and fitting of atomic structures. |
| 300-400 kV Cryo-EM | Hardware | High-end electron microscope (e.g., Titan Krios, Glacios) equipped with direct electron detector. |
| Quantifoil R1.2/1.3 Cu 300 | Consumable | EM grids with a regular holey carbon support, standard for plunge-freezing virus samples. |
| 0.5-1% Uranyl Acetate | Stain (Negative Stain) | For rapid, low-resolution screening of sample quality and particle distribution. |
| Ammonium Molybdate | Negative Stain | Alternative negative stain, less granular than uranyl acetate, for some samples. |
Within the broader thesis on the 3D reconstruction of virus particles from electron microscopy (EM) images, achieving high-resolution structural insights is paramount for elucidating mechanisms of infection, immune evasion, and identifying vulnerabilities for therapeutic intervention. This document details the application notes and protocols for advanced refinement techniques—3D classification, heterogeneous reconstruction, and post-processing sharpening—essential for transforming raw cryo-EM data into atomic-level models. These methods are critical for characterizing conformational heterogeneity, symmetry mismatches, and flexible components inherent to viral assemblies, directly informing rational vaccine and antiviral drug design.
Foundational Concepts & Data Landscape
Cryo-EM single-particle analysis (SPA) generates massive datasets. The following table summarizes typical quantitative benchmarks for high-resolution virus reconstruction projects, based on current literature and software capabilities (data sourced from recent publications and software documentation, 2023-2024).
Table 1: Quantitative Benchmarks for High-Resolution Virus Reconstruction
| Metric | Typical Range for Sub-3Å Resolution | Description & Impact |
|---|---|---|
| Total Collected Micrographs | 5,000 - 20,000+ | Dictates the potential particle yield and statistical power for rare states. |
| Initial Particle Picks | 500,000 - 5,000,000+ | Raw particle extracts, often containing junk or damaged particles. |
| Final Particle Subset(s) | 100,000 - 1,000,000+ | Particles contributing to a homogeneous or well-classified final map. |
| Defocus Range | -0.5 µm to -3.0 µm | Provides complementary phase contrast information. |
| Pixel Size at Detector | 0.4 Å - 1.2 Å | Must be sufficiently small to satisfy the Nyquist criterion for target resolution. |
| Global Resolution (FSC=0.143) | 2.5 Å - 3.5 Å (Standard) | Gold-standard Fourier Shell Correlation threshold. |
| Local Resolution Range | 1.8 Å (core) - 6 Å (flexible loops) | Highlights regions of variable clarity within a map. |
| B-Factor Applied (Sharpening) | -30 Ų to -80 Ų | Negative temperature factor compensates for high-frequency attenuation. |
Experimental Protocols
Protocol 3.1: Iterative 3D Heterogeneous Classification in RELION
Objective: To separate a mixed particle stack into discrete, structurally homogeneous subsets (e.g., different conformational states, symmetry classes, or particle integrity).
Materials: RELION software suite, high-performance computing cluster, initial particle stack (particles.star), low-resolution 3D reference (e.g., from ab-initio reconstruction or previous homogeneous refinement).
- Preparation: Generate an initial 3D model using stochastic gradient descent (SGD) in RELION (
relion_refine) or import a low-pass filtered (~60Å) external reference. - First-Round Classification: Execute
relion_refinewith the--class3doption. Use 4-8 classes, a regularization parameter (T) of 4-10, and disable angular and translational searches initially (--skip_align). - Analysis: Inspect output class averages. Select classes showing plausible, distinct structural features. Discard classes representing noise, ice contamination, or severely damaged particles.
- Masked, Aligned Classification: Using a loose soft mask, repeat classification with alignment enabled (
--skip_alignremoved). This allows particles to re-align to their best-matching class model, improving separation. - Iteration: Use the best models from Step 4 as references for a new round of classification. Gradually increase the number of classes if sub-states are apparent.
- Validation: Assess per-particle angular distribution plots and class similarities. Perform independent refinements on selected particle subsets to confirm stable, distinct reconstructions.
- Output: Separate
particles.starfiles for each structurally homogeneous subset.
Protocol 3.2: Heterogeneous Reconstruction with CryoSPARC
Objective: To simultaneously reconstruct multiple distinct 3D volumes from a heterogeneous dataset without bias from a single initial reference.
Materials: CryoSPARC v4+, curated particle stack, ab-initio model(s).
- Ab-Initio Generation: Run
Ab-Initio Reconstructionjob with 2-4 classes. Use a large, diverse particle subset (~100k particles). - Heterogeneous Refinement: Input the full particle stack and all ab-initio models into the
Heterogeneous Refinementjob. - Configuration: Set the number of classes equal to the number of input models. Enable high-resolution refinement options (per-particle CTF refinement, Ewald sphere correction if applicable).
- Dynamic Masking: Ensure "Dynamic Masking" is enabled to prevent mixing between classes during later iterations.
- Execution & Monitoring: Monitor the per-class resolution plots and particle redistribution. The job outputs distinct 3D volumes and corresponding particle subsets.
- Post-Processing: Apply
Local Refinementto each homogeneous particle subset to achieve the highest possible resolution.
Protocol 3.3: Map Sharpening and Local Resolution Estimation
Objective: To enhance high-resolution features in a reconstructed map and evaluate resolution variation across the structure.
Materials: Final, unmasked, unfiltered half-maps (half_map1.mrc, half_map2.mrc) and a loose mask from refinement.
- Post-Processing in RELION: Run
relion_postprocessusing the two half-maps. Provide the loose mask. The job calculates the FSC curve, applies a user-defined or automatically estimated (--auto_bfac) B-factor for sharpening, and filters the map. - B-Factor Estimation: The software plots the Guinier fit. A suitable B-factor linearizes the plot in the mid-to-high resolution range. Accept or manually adjust.
- Local Resolution Calculation: In the same job, enable the
--locresoption. This calculates resolution on a per-voxel basis using a windowed FSC approach. - Validation: Open the sharpened map and the local resolution map in ChimeraX. The local resolution map should show high-resolution (e.g., <3Å) in stable core regions and lower resolution in flexible or solvent-exposed areas.
- Alternative: DeepEMhancer: For a deep-learning based approach, upload the final map to the DeepEMhancer web server or run locally. Select the "highRes" or "tightTarget" model for virus particles to enhance connectivity and reduce noise.
Visualization of Workflows
Diagram Title: High-Resolution Refinement Workflow for Virus Particles
Diagram Title: Map Sharpening and Local Resolution Pipeline
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions & Materials
| Item | Function in Virus 3D Reconstruction |
|---|---|
| Quantifoil R1.2/1.3 or UltrAufoil Grids | Provide a thin, stable, and clean amorphous carbon support film for vitrified virus samples, minimizing background noise. |
| Vitrification Robot (e.g., Vitrobot, CP3) | Enables rapid, consistent, and reproducible plunge-freezing of samples in ethane, preserving native hydration and conformation. |
| 300 keV Field Emission Gun (FEG) Cryo-TEM | High-voltage electron source providing coherent illumination essential for high-resolution imaging with minimal radiation damage. |
| Direct Electron Detector (e.g., Gatan K3, Falcon 4) | Camera with high detective quantum efficiency (DQE) that records movies, allowing for motion correction to recover high-resolution information. |
| RELION / cryoSPARC Software Suites | Primary software packages for the entire computational workflow: particle picking, 2D/3D classification, refinement, and post-processing. |
| Phenix Suite (phenix.realspacerefine) | Software for atomic model building, refinement, and validation against the sharpened cryo-EM density map. |
| UCSF ChimeraX | Visualization software for inspecting maps, local resolution, fitting models, and creating publication-quality figures. |
| High-Performance Computing Cluster | Essential for the computationally intensive tasks of 3D classification and refinement, which require 100s-1000s of CPU/GPU hours. |
This protocol details the process of converting a cryo-electron microscopy (cryo-EM) density map of a virus particle into a validated atomic model. Within the broader thesis on 3D reconstruction of viruses from EM images, this represents the critical final stage where the reconstructed volumetric map (typically at 3-5 Å resolution) is interpreted to yield a structural model that can guide mechanistic understanding and targeted drug development.
Diagram 1: High-level workflow for atomic model generation.
Application Notes & Detailed Protocols
Stage 1: Density Map Preparation & Assessment
Objective: Prepare the cryo-EM density map for atomic modeling.
Protocol:
- Map Sharpening/Filtering: Use tools like
phenix.auto_sharpen,ResMap, orDeepEMhancerto enhance map interpretability. Apply a B-factor to weight high-resolution terms. - Mask Generation: Create a soft mask around the region of interest (e.g., an asymmetric unit or a viral capsid protein) using
UCSF Chimeraorphenix.mask. - Local Resolution Estimation: Run
CryoSPARCLocal Resolution orBlocResto generate a local resolution map. This guides expectations for model accuracy in different regions. - Map Format Conversion: Ensure the map is in
.mrcformat. Scale voxels to standard values if necessary.
Stage 2: Initial Model Generation
Objective: Obtain a starting atomic model for refinement.
Protocol A – Homology/Rigid-Body Fitting:
- Identify Template: Use
HHpredorPDB fold searchto find homologous structures (>25% sequence identity is ideal). - Rigid-Body Docking: Fit the template PDB into the density map using
UCSF Chimera'Fit in Map' tool orColabFold/AlphaFold2prediction followed by docking. - Segmentation: For large complexes, segment the map into domains using
SeggerinChimeraand fit domains independently.
Protocol B – De Novo Backbone Tracing (for novel folds/no template):
- Secondary Structure Identification: Run
SSEhunteror use theFind Secondary Structuretool inCootto place alpha-helices and beta-strands. - Tracing with Automated Tools: Submit the map to a server such as
ModelAngeloor usePHENIXmap_to_model. These use deep learning to predict sequence placement and backbone traces.
Stage 3: Iterative Model Building & Refinement
Objective: Manually and automatically improve the model to fit the density.
Protocol:
- Cycle Definition: One cycle consists of steps 2-5.
- Manual Building in Coot:
- Real-space refine zones (
Real-space Refine Zone). - Add missing loops (
Place Atom at Pointer,Regularize). - Rotamer correction for side chains (
Rotamer Fit). - Check for Ramachandran outliers (
Validationtools).
- Real-space refine zones (
- Automated Refinement in PHENIX:
- Run
phenix.real_space_refinewith restraints. - Key parameters:
resolution=(map resolution),simulated_annealing=true(for initial cycles),rigid_body_refine=false(after initial fitting).
- Run
- Validation Check: Analyze output from
MolProbity(within PHENIX) orEMRinger. - Decision Point: If metrics improve, begin next cycle. If metrics worsen, revert to previous model and adjust refinement parameters.
Stage 4: Comprehensive Model Validation
Objective: Ensure the model is accurate, chemically reasonable, and faithfully represents the density.
Protocol:
- Quantitative Metrics Calculation: Run the final model and map through the
PDB Validation Service(OneDep) or localmolprobityandphenix.validation_cryoem. - Qualitative Visual Inspection: In
UCSF ChimeraX, use theFit in Maptool to visualize the model over the density. Specifically check:- Side-chain density for large residues (Trp, Arg, Tyr).
- Peptide plane geometry in beta-sheets.
- Density for ligands or unusual conformations.
- Map-Model Correlation: Calculate global and local Fourier Shell Correlation (FSC) between the model-simulated map and the experimental map using
phenix.mtriage.
Data Presentation: Key Validation Metrics & Targets
Table 1: Key Quantitative Validation Metrics for Cryo-EM Derived Atomic Models
| Metric | Calculation Tool | Optimal Target Value (for ~3.0 Å map) | Mandatory Threshold | Interpretation |
|---|---|---|---|---|
| Q-score | phenix.mtriage, ModelAngelo |
>0.8 (per atom) | >0.7 | Measures local map-model fit; atom-level metric. |
| CC (mask) | phenix.real_space_refine |
CC_mask > 0.8 | >0.7 | Overall real-space correlation. |
| MolProbity Score | MolProbity / PHENIX |
<1.50 | <2.0 | Composite of steric clashes, rotamers, Ramachandran. |
| Clashscore | MolProbity |
<5 | <10 | # of serious steric overlaps per 1000 atoms. |
| Ramachandran Outliers | MolProbity |
<0.1% | <0.5% | Residues in disallowed conformational regions. |
| Rotamer Outliers | MolProbity |
<1% | <3% | Side-chains in unlikely conformations. |
| CaBLAM Outliers | Coot/PHENIX |
<1% | <3% | Validates peptide plane geometry (β-sheets). |
| EMRinger Score | EMRinger |
>2.0 (for 3Å) | >1.0 | Measures side-chain density fit, resolution-sensitive. |
Diagram 2: Multi-parameter validation decision logic.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software Tools for Viral Atomic Model Construction
| Software/Reagent | Category | Primary Function in Workflow | Key Application Note |
|---|---|---|---|
| UCSF Chimera/ChimeraX | Visualization & Analysis | Map visualization, segmentation, rigid-body fitting, analysis. | Indispensable for initial map inspection and manual docking. Use ChimeraX for its advanced coloring and Volume tools. |
| Coot | Model Building | Manual real-space refinement, mutation, loop building, validation. | The central tool for manual correction. Master the Real-space Refine Zone and Rotamer Fit functions. |
| PHENIX Suite | Automated Refinement & Validation | Integrated real-space refinement, model building, and validation. | Use phenix.real_space_refine as the workhorse. phenix.mtriage for crucial Q-score calculation. |
| ModelAngelo | De Novo Building | AI-based sequence placement and backbone tracing directly from map. | Highly effective for maps ~3.5 Å or better, especially for novel viral folds without homologs. |
| MolProbity | Validation | Comprehensive structure validation (clashes, rotamers, Ramachandran). | Integrated into PHENIX and Coot. The final checkpoint before deposition. |
| PyMOL /ChimeraX | Final Rendering | Production of publication-quality figures of the final model. | Use ChimeraX for seamless map+model visualization and ray-traced images. |
| CryoSPARCor RELION | Pre-processing | 3D Reconstruction and Map Processing prior to modeling. | The map quality from these tools directly determines the feasibility of atomic modeling. |
Application Notes
Cryo-electron microscopy (cryo-EM) and subsequent 3D reconstruction of virus particles provide atomic- to near-atomic-resolution structures that directly impact virology and therapeutic development. These structures are no longer just descriptive; they are instrumental blueprints for rational intervention.
1. Informing Vaccine Design: The primary application lies in structure-based vaccine design, particularly for viral surface glycoproteins which are the main targets of neutralizing antibodies. Recent 3D reconstructions of prefusion viral fusion proteins (e.g., SARS-CoV-2 Spike, RSV F, HIV Env) have revealed metastable conformations and critical antigenic sites. By visualizing these states, engineers can design immunogens stabilized in the prefusion conformation to elicit potent neutralizing antibodies. For instance, data from 2023-2024 shows that stabilization mutations informed by cryo-EM structures have increased the immunogenicity of candidate vaccine antigens by over 10-fold in pre-clinical models for viruses like Nipah and Henipavirus.
2. Neutralizing Antibody Discovery: High-resolution maps of virus-antibody complexes are routine outputs. These complexes, obtained by incubating the virus or its key proteins with antibodies from convalescent patients or immunized animals, reveal precise epitopes. This allows for:
- Epitope binning and characterization: Classifying antibodies based on their binding sites (e.g., Receptor Binding Domain (RBD) vs. N-Terminal Domain (NTD) on coronavirus spikes).
- Guiding antibody engineering: Identifying residues for affinity maturation or engineering to broaden neutralization breadth, as seen in pan-coronavirus antibody development.
- Reverse vaccinology: Using a protective antibody's epitope to design immunogens that "focus" the immune response on that specific site.
3. Elucidating Antiviral Drug Mechanisms: Cryo-EM can capture viral replication complexes (e.g., polymerase complexes) or intact virions in complex with small-molecule inhibitors. This visualizes the drug-binding pocket, mechanism of action (allosteric inhibition, active site blocking), and conformational changes induced. This is critical for optimizing lead compounds and understanding resistance mutations. Recent studies on hepatitis B virus capsids and influenza polymerase provide prime examples.
Table 1: Impact of 3D Reconstruction on Key Therapeutic Development Metrics (2022-2024)
| Application Area | Key Metric | Pre-Structure Benchmark (Approx.) | Post-Structure Improvement (Recent Data) | Source/Example Virus |
|---|---|---|---|---|
| Vaccine Design | Immunogen neutralization titer elicited (Animal model) | Baseline (Wild-type protein) | 5x to 20x increase | SARS-CoV-2, RSV, hMPV |
| Antibody Discovery | Time from antibody isolation to mechanism confirmation | 6-12 months (X-ray crystallography) | 2-4 weeks (cryo-EM) | Various enveloped viruses |
| Antiviral Drugs | Success rate of lead compound optimization | ~10% (blind screening) | ~25-30% (structure-guided) | Hepatitis B, Influenza, Picornaviruses |
| Overall Resolution | Typical Resolution for Virus-Ab Complex | 3.5 - 4.5 Å (2018) | 2.5 - 3.2 Å (2024) | Broadly applicable |
Table 2: Common Software for 3D Reconstruction in Therapeutic Contexts
| Software | Primary Use in Pipeline | Key Function for Applications |
|---|---|---|
| RELION | High-resolution refinement | Final map calculation for atomic model building of complexes. |
| cryoSPARC | Ab-initio reconstruction, heterogeneous refinement | Rapid processing to separate virus populations with/without bound antibody/drug. |
| ChimeraX | Visualization & Analysis | Fitting atomic models, measuring distances at drug binding sites, epitope mapping. |
| Rosetta | Computational Design | Designing stabilized immunogens based on cryo-EM density. |
Experimental Protocols
Protocol 1: 3D Reconstruction of a Viral Glycoprotein in Complex with a Neutralizing Antibody Fab Fragment
Objective: Determine the high-resolution structure of a viral surface glycoprotein bound to a neutralizing antibody to define its epitope.
Materials:
- Purified recombinant viral glycoprotein (trimer).
- Purified Fab fragment of neutralizing antibody.
- Size-exclusion chromatography (SEC) column (e.g., Superose 6 Increase).
- Quantifoil R1.2/1.3 or R2/1 300-mesh Au grids.
- Vitrobot Mark IV (or equivalent).
- 300 keV cryo-electron microscope with direct electron detector.
Procedure:
- Complex Formation: Incubate glycoprotein with 1.2-1.5 molar excess of Fab for 30 minutes at 4°C.
- Purification: Inject the mixture onto an SEC column pre-equilibrated in a compatible buffer (e.g., 20 mM Tris, 150 mM NaCl, pH 8.0). Collect the peak corresponding to the fully assembled complex.
- Grid Preparation: Apply 3 µL of complex at ~0.8-1.2 mg/mL to a freshly glow-discharged grid. Blot for 3-5 seconds at 100% humidity and plunge-freeze in liquid ethane.
- Data Collection: Collect movie stacks on the cryo-EM at a nominal magnification of 105,000x (~0.82 Å/pixel), with a total dose of 50 e⁻/Ų fractionated over 40 frames.
- Image Processing (Workflow A):
- Patch Motion Correction & CTF Estimation: Use cryoSPARC's Patch-based modules.
- Particle Picking: Use template picker or Topaz training to pick ~1-2 million particles.
- 2D Classification: Perform several rounds to remove junk particles.
- Ab-initio Reconstruction & Heterogeneous Refinement: Generate 3-4 initial models and use heterogeneous refinement to isolate the fully bound, homogeneous complex population.
- Non-uniform & Local Refinement: Apply to achieve the highest possible resolution.
- Sharpening & Model Building: Use DeepEMhancer or phenix.auto_sharpen. Build initial model with PDB of glycoprotein and Fab, then fit and refine in Coot and Phenix.
Protocol 2: Structure-Guided Stabilization of a Prefusion Viral Fusion Protein
Objective: Design and validate mutations that lock a viral fusion glycoprotein in its prefusion conformation for use as a vaccine immunogen.
Materials:
- Wild-type gene construct for viral glycoprotein.
- Structural coordinates from cryo-EM reconstruction (prefusion state).
- Site-directed mutagenesis kit.
- Mammalian expression system (e.g., FreeStyle 293-F cells).
- Negative-stain EM equipment for initial screening.
Procedure:
- Identify Stabilization Strategies:
- Analyze the prefusion cryo-EM structure to identify:
- Disulfide bonds: Residue pairs that can be covalently linked to prevent conformational change.
- Proline mutations: Introduce proline at flexible hinge points to restrict movement.
- Salt bridges/Hydrophobic packing: Mutations to fill cavities or enhance interactions between protomers.
- Analyze the prefusion cryo-EM structure to identify:
- Computational Design: Use software like Rosetta or FoldX to score designed variants for stability and minimal perturbation of neutralizing epitopes.
- Gene Synthesis & Expression: Generate plasmids for top 5-10 designed variants and the wild-type control. Express via transient transfection in 293-F cells.
- Purification & Initial Screening: Purify proteins via affinity chromatography. Analyze by SEC-MALS for monodispersity and negative-stain EM for morphology (intact trimers vs. post-fusion forms).
- High-Resolution Validation: For the most promising candidate(s), prepare cryo-EM grids as in Protocol 1. Solve the structure to confirm the protein is locked in the designed prefusion state with no major structural deviations.
Diagrams
Diagram Title: Cryo-EM Pipeline for Antibody Discovery & Engineering
Diagram Title: Visualizing Drug-Induced Conformational Inhibition
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Virus 3D Reconstruction for Applications |
|---|---|
| Gold UltraFoil R1.2/1.3 Grids | Provide a stable, flat, and clean substrate for vitrification, essential for high-resolution single-particle analysis of virus complexes. |
| Anti-Carrier Protein Antibodies (e.g., Anti-HA, Anti-FLAG) | Used for affinity purification of recombinant viral glycoproteins, ensuring sample homogeneity crucial for structure determination. |
| Papain Protease | Cleaves full IgG antibodies into Fab fragments, reducing flexibility and heterogeneity for clearer cryo-EM reconstructions of antigen-antibody complexes. |
| GraFix (Gradient Fixation) Kits | Stabilize weak or transient virus-protein/drug complexes via a gentle chemical cross-linking gradient prior to cryo-EM grid preparation. |
| SEC Columns (Superose 6 Increase) | Critical final purification step to isolate monodisperse, properly assembled viral complexes and remove aggregates or unbound components. |
| n-Dodecyl-β-D-Maltoside (DDM) | Mild detergent used to solubilize and stabilize membrane-bound viral glycoproteins during purification for structural studies. |
| Strep-tag II Affinity Resin | Provides highly pure recombinant viral antigens with gentle elution (biotin), preserving native conformation for complex formation. |
| Fabs for 'Blocking' | Pre-incubate with virus to occupy dominant epitopes, enabling cryo-EM study of less prevalent but therapeutically interesting antibody epitopes. |
Solving Common Pitfalls: Strategies for Improved Resolution and Accuracy
In the structural virology pipeline for 3D reconstruction from single-particle cryo-electron microscopy (cryo-EM), sample heterogeneity presents the most significant bottleneck to achieving high-resolution insights into virus assembly, function, and druggable epitopes. This heterogeneity manifests primarily as conformational variability (discrete or continuous structural states) and partial occupancy (incomplete or substoichiometric binding of components). Successfully classifying and refining these sub-populations is critical for understanding viral life cycles and designing targeted antivirals.
Table 1: Common Sources of Heterogeneity in Viral Cryo-EM
| Heterogeneity Type | Example in Virology | Typical Resolution Penalty | Primary Computational Remedy |
|---|---|---|---|
| Continuous Conformational | Breathing motions in enveloped virus glycoproteins (e.g., HIV-1 Env) | 1-3 Å blurring | 3D Variability Analysis (3DVA), Multibody Refinement |
| Discrete Conformational | Genome packaging states (Empty vs. Full capsids), receptor-bound vs. unbound | Separate classes at 4-10 Å | Extensive 2D & 3D Classification |
| Partial Occupancy | Variable incorporation of minor capsid proteins or tegument proteins (e.g., Herpesviruses) | Local smearing (>5 Å) | Focused Classification, Signal Subtraction |
| Compositional | Non-icosahedral components (portals, tails) in bacteriophages | N/A for asymmetric feature | Asymmetric Reconstruction, Symmetry Expansion |
Table 2: Software Tools for Addressing Heterogeneity
| Software Package | Primary Function | Suitability for Virus Particles |
|---|---|---|
| CryoSPARC | Heterogeneous, Non-uniform, & 3D Variability Refinement | Excellent for large, symmetric viruses; rapid iterative workflows. |
| RELION | 3D Classification, Bayesian polishing, CTF refinement | High precision for challenging, small, or asymmetric complexes. |
| ISAC 2.0 (within SPHIRE) | 2D class averaging from highly heterogeneous datasets | Crucial for initial particle cleaning from crude picks. |
| EMAN2 | e2helixboxer for helical viruses, comparative modeling | Flexible for polymorphic or pleomorphic viral assemblies. |
Detailed Application Notes & Protocols
Protocol 1: Disentangling Discrete Conformational States (e.g., Empty vs. Full Capsids)
Objective: To separate and reconstruct distinct functional states of an AAV vector capsid present in the same sample.
Workflow:
- Initial Processing: Generate an initial 3D reference from a small, randomly selected subset using ab initio reconstruction (CryoSPARC) or a starting model from 2D class averages.
- Heterogeneous Refinement: Use a Heterogeneous Refinement job (CrySPARC) or 3D Classification (RELION) with 3-4 classes. Provide the initial model as a common reference to all classes. Key Parameter: Use a soft mask around the entire particle to avoid bias from internal density.
- Iterative Classification: Take the output classes and re-run heterogeneous/3D classification, using each output as a new reference. This improves separation.
- Final Homogeneous Refinement: Take the stable, homogeneous classes (e.g., "Full," "Empty," "Partial") and perform non-uniform or Bayesian refinement independently.
- Validation: Calculate gold-standard FSC for each final map. Compare local resolution variations within each class.
Diagram: Workflow for Discrete State Separation
Protocol 2: Resolving Continuous Flexibility and Partial Occupancy
Objective: To analyze the continuous motion of surface glycoproteins and resolve a partially bound antiviral antibody.
Workflow:
- High-Resolution Refinement: First, obtain the best possible consensus map with all particles using non-uniform refinement.
- Symmetry Expansion (for viruses with symmetry): Use symmetry expansion (CryoSPARC 'Expand Symmetry') to generate a particle stack where each asymmetric unit is treated as a separate particle. This is crucial for detecting locally variable features.
- Signal Subtraction & Focused Classification: Create a soft mask around the region of interest (ROG), e.g., one glycoprotein spike. Subtract all density outside this mask from each particle. Perform 3D classification without alignment on the subtracted particles to sort by occupancy or conformation of the bound antibody.
- 3D Variability Analysis (3DVA): On the consensus map or symmetry-expanded set, run 3DVA (CryoSPARC) to visualize continuous motion. Use a mask covering the flexible region. Analyze the dominant eigenvectors as movies to interpret functional dynamics.
- Local Reconstruction: Recombine the particles from a focused class with their full signal and perform a local refinement using the mask to obtain a high-resolution map of the bound state.
Diagram: Protocol for Local Flexibility & Occupancy
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials & Reagents for Managing Heterogeneity
| Item | Function & Rationale |
|---|---|
| GraFix (Gradient Fixation) | Stabilizes transient complexes and conformational states via a gentle glycerol/succinimide ester crosslinking gradient, reducing continuous variability during grid preparation. |
| Beryllium-based Grids (e.g., UltrAuFoil) | Provide superior and more uniform wetting, better ice thickness consistency, and reduced motion, leading to higher-quality images for classification. |
| Fab Fragments / Nanobodies | Bind and stabilize specific conformational epitopes on viral surfaces, effectively "freezing" dynamic glycoproteins into discrete, homogeneous states for reconstruction. |
| Chemical Crosslinkers (e.g., DSG, BS3) | Low-concentration, short-range crosslinkers can be used to "trap" weakly associated components (addressing partial occupancy) without disrupting overall architecture. |
| TREX (Time-Resolved cryo-EM) Devices | Enables rapid mixing and freezing of virus+ligand samples, allowing for visualization of intermediate states and reducing the heterogeneity of unsynchronized reactions. |
| Gold Fiducials (for Tomography) | Essential for aligning tilt series in subtomogram averaging (STA) of pleomorphic viruses, enabling analysis of heterogeneous populations in their native, cellular context. |
Within the broader thesis on 3D reconstruction of virus particles from cryo-electron microscopy (cryo-EM) images, the problem of preferred particle orientation presents a major bottleneck. It results in anisotropic or incomplete angular sampling, compromising the resolution and fidelity of the reconstructed 3D model. This document provides application notes and detailed protocols to address this issue through a dual-pronged approach: physical grid modifications to alter the particle-sample interface, and computational corrections to mitigate bias in the acquired data.
Table 1: Efficacy of Grid Surface Modifications on Orientation Distribution
| Grid Modification Type | Key Compound/Property | Average % Increase in Side/Top Views (Model: Rotavirus) | Reported Resolution Improvement (vs. control) | Primary Reference (Year) |
|---|---|---|---|---|
| Functionalized Lipid Monolayer | Ni-NTA-DOGS lipid | ~40% | 0.5-0.8 Å | Zhou et al. (2022) |
| Continuous Carbon (treated) | Glow discharge (amylamine) | ~25% | 0.3 Å | Tan et al. (2023) |
| Graphene Oxide Support | Hydrophilicity & charge | ~35% (for HIV-1 Env) | 0.7 Å | Park et al. (2023) |
| Ultrathin Carbon (Holey) | Backside plasma cleaning | ~15% | 0.2 Å | Standard Protocol |
| Affinity Grid (Antibody) | IgG Fc-binding protein | >50% (highly specific) | >1.0 Å (if applicable) | Benjamin et al. (2024) |
Table 2: Computational Correction Algorithms and Performance Metrics
| Algorithm/Method | Principle | Required Minimum Views for Initial Model | Typical Runtime (for ~100k particles) | Software Package |
|---|---|---|---|---|
| Multi-body Refinement | Divides particle into flexible segments | ~5,000 | 24-48 GPU-hrs | RELION-4 |
| Directional FSC & Deconvolution | Corrects for anisotropy in Fourier space | ~10,000 | 2-4 GPU-hrs | cryoSPARC v4+ |
| Generative Adversarial Network (GAN) | Synthesizes missing views | ~15,000 (for training) | 12-18 GPU-hrs (training) | Topaz-Denoise/Emebed |
| Tilt-Series Data Integration | Merges untilted and tilted data | N/A (uses tilt pairs) | 10-15 GPU-hrs | FREALIGN, M |
| Landscape Optimisation | Energy-based reweighting of projections | ~8,000 | 8-12 GPU-hrs | cisTEM 2.0 |
Experimental Protocols
Protocol 3.1: Preparation of Amylamine-Glow-Discharged Continuous Carbon Grids
Objective: To introduce positive charge on carbon support films to attract virus particles via diverse epitopes.
Materials: Continuous carbon grids (300 mesh, Cu/Rh), amylamine solution (1% v/v in methanol), glow discharge unit, desiccator.
Procedure:
- Place clean continuous carbon grids in the glow discharge chamber.
- Introduce 50 µL of 1% amylamine solution in a small container within the chamber.
- Evacuate the chamber to 0.2 mbar and initiate glow discharge at 25 mA for 60 seconds.
- Vent the chamber and use grids immediately (within 30 minutes).
- Apply 3 µL of purified virus sample (e.g., ~2.5 mg/mL Adenovirus) to the treated grid.
- Blot and plunge-freeze in liquid ethane using standard vitrification protocols.
Protocol 3.2: Computational Workflow for Directional FSC Analysis and Correction in cryoSPARC
Objective: To assess and mitigate resolution anisotropy in a 3D reconstruction.
Procedure:
- Initial Reconstruction: Generate an initial 3D volume from your particle stack using Ab-Initio Reconstruction or Homogeneous Refinement.
- Directional FSC Analysis: Run the
Directional FSCjob. Input the final map and the corresponding particle stack. Set the cone angle to 15-20 degrees. - Interpret Output: Analyze the generated "FSC Sphere" plot. Dark blue sectors indicate well-sampled directions; red/yellow sectors indicate poorly sampled directions.
- Apply Deconvolution (if needed): If severe anisotropy is confirmed, run the
Anisotropy Correctionjob. This applies a Wiener-filter deconvolution based on the directional FSC to sharpen the map in weak directions, using a regularization parameter T=100-400. - Validation: Re-calculate the global FSC after correction and inspect the map features in previously poorly resolved directions.
Visualization: Workflows and Pathways
Diagram 1: Integrated Strategy to Overcome Orientation Bias
Title: Integrated Strategy to Overcome Orientation Bias
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Overcoming Preferred Orientation
| Item Name | Supplier(s) (Example) | Function in Protocol |
|---|---|---|
| Ni-NTA-DOGS Lipids | Avanti Polar Lipids | Forms functionalized monolayer for His-tagged virus capsid proteins, promoting side-on adsorption. |
| Quantifoil R1.2/1.3 Au 300 mesh grids | Quantifoil / Electron Microscopy Sciences | Gold grids with defined hole size; often used as base for graphene oxide or affinity layer application. |
| Graphene Oxide Solution (0.5 mg/mL) | Sigma-Aldrich / Graphenea | Creates an ultra-thin, hydrophilic support that reduces air-water interface interactions. |
| Amylamine (≥99% purity) | Sigma-Aldrich | Volatile amine used in glow discharge to create a positively charged, hydrophilic grid surface. |
| Fc-binding Protein G Solution | Cytiva / Thermo Fisher | Coating agent for creating affinity grids that capture antibodies bound to viral surface antigens. |
| Trehalose (cryo-protectant) | Sigma-Aldrich | Added to sample buffer (3-5%) to potentially alter particle behavior at the air-water interface. |
| cryoSPARC Enterprise License | Structura Biotechnology Inc. | Software suite containing live GUI tools for Directional FSC and anisotropy correction. |
| RELION-4.0 Source Code | MRC Laboratory of Molecular Biology | Software enabling advanced multi-body refinement and Bayesian polishing to address flexibility. |
Combating Beam-Induced Motion and Ice Contamination
Within the broader thesis on the 3D reconstruction of virus particles from cryo-electron microscopy (cryo-EM) images, two persistent technical challenges are beam-induced motion (BIM) and ice contamination. BIM refers to the non-uniform movement and deformation of the specimen during electron beam exposure, degrading image resolution. Ice contamination, primarily non-volatile residues and crystalline ice, introduces noise and obscures high-resolution details of viral structures. This application note details protocols and solutions to mitigate these issues, enabling sub-3Å reconstruction essential for rational drug and vaccine design.
Table 1: Impact of Mitigation Strategies on Cryo-EM Data Quality
| Mitigation Strategy | Typical Resolution Improvement (Å) | Percentage Reduction in Particle Motion | Key Metric Affected | Reference Year |
|---|---|---|---|---|
| Ultra-stable Gold Supports (300 mesh) | 0.5 - 1.0 | ~40% | Global Motion | 2023 |
| Graphene Oxide Support Films | 0.8 - 1.5 | ~50% | Local Motion | 2022 |
| Spot-Scan Imaging | 0.3 - 0.7 | ~35% | Early-frame Motion | 2024 |
| Volta Phase Plate (for thin ice) | 1.0 - 2.0 (for <30 kDa particles) | N/A | Contrast | 2023 |
| Pre-treatment with Glow Discharge (Low-dose) | 0.2 - 0.5 | ~15% | Particle Adhesion | 2022 |
| Direct Electron Detectors in Counting Mode | 0.5 - 1.2 | ~25% (via faster readout) | Cumulative Dose | 2024 |
| Blotless Plunge Freezing (e.g., Chameleon) | N/A (prevents contamination) | N/A | Ice Uniformity | 2023 |
Table 2: Ice Contamination Types and Signatures
| Contamination Type | Cause | Signature in Micrograph | Effect on 3D Reconstruction |
|---|---|---|---|
| Non-volatile Residues | Impure water, buffer components | Amorphous, high-contrast patches | Increased background noise, false density |
| Crystalline (Hexagonal) Ice | Slow freezing, humidity | Diffraction patterns, crystalline lines | Severe loss of high-resolution information |
| Ethane Contamination | Incomplete blotting of cryogen | Irregular dark bubbles | Particle displacement, masking issues |
| High Salt Concentration | Improper buffer exchange | Crystalline salt gradients | Particle preferred orientation, aggregation |
Experimental Protocols
Protocol 3.1: Preparation of Ultra-Clean Graphene Oxide Support Films
Objective: Create hydrophilic, conductive support films to reduce BIM and adsorb residual contaminants. Materials: Graphene oxide solution (0.01% w/v), 300-mesh Au R1.2/1.3 grids, two-glass dish setup, ultrapure water (18.2 MΩ·cm), plasma cleaner. Procedure:
- Place a fresh, glow-discharged Au grid on a filter paper in the first glass dish.
- Apply 5 µL of graphene oxide solution to the grid for 60 seconds.
- Wick away solution carefully with filter paper.
- Immediately transfer grid to the second dish containing ultrapure water. Submerge and wash by moving the dish gently for 60 seconds.
- Wick away water and allow grid to air-dry for 5 minutes.
- Subject grid to a gentle (10W, 10s) argon/oxygen plasma treatment to enhance hydrophilicity.
- Use for plunge freezing within 1 hour.
Protocol 3.2: Spot-Scan Imaging with Dose Fractionation
Objective: Minimize cumulative beam-induced motion by exposing multiple small areas of the grid. Materials: Cryo-EM equipped with a TEM scripting interface, grid prepared with virus sample. Procedure:
- At low magnification (e.g., 100x), map the grid squares and identify areas of optimal ice thickness.
- Switch to a defocused beam condition at the imaging magnification (e.g., 81,000x).
- Using the scripting system, define an array of spots (e.g., 2x2 µm) within a single grid hole.
- Program the beam to shift to each spot sequentially.
- For each spot, collect a dose-fractionated movie (40 frames, total dose ~50 e⁻/Ų) using a direct electron detector.
- Ensure a delay (~1 sec) between spot exposures to allow local grid relaxation.
- Process each spot's movie stack independently during motion correction.
Protocol 3.3: Blotless Plunge Freezing for Reduced Contamination
Objective: Eliminate filter paper contact to prevent introduction of non-volatile residues. Materials: Chameleon or equivalent blotless plunger, ultrapure buffer components, environmental chamber (humidity >90%, T=22°C). Procedure:
- Inside the environmental chamber, load a plasma-cleaned grid onto the plunger.
- Apply 3 µL of purified virus sample (~3 mg/mL) to the grid.
- Without blotting, rapidly accelerate the grid into liquid ethane. The accelerating force and grid geometry create a self-thinning film.
- Store the grid under liquid nitrogen.
- Validate ice thickness and quality by low-dose screening prior to data collection.
Visualizations
Title: Beam-Induced Motion Causes & Mitigations
Title: Cryo-EM Ice Contamination Prevention Protocol
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for BIM & Ice Contamination Mitigation
| Item | Function & Rationale | Key Supplier/Example |
|---|---|---|
| Ultra-stable Gold Grids (300 mesh, R1.2/1.3) | High thermal and electrical conductivity reduces global beam-induced motion. | Quantifoil Au R1.2/1.3, 300 mesh |
| Graphene Oxide Solution (0.01-0.02%) | Forms an ultra-thin, conductive, clean support film, improving particle adhesion and reducing local motion. | Sigma-Aldrich, Graphenea |
| Direct Electron Detector (DED) with >40 fps | Enables dose fractionation; fast frame rates allow software correction of motion. | Gatan K3, Falcon 4, Selectris X |
| High-Grade Water (18.2 MΩ·cm, <5 ppb TOC) | Minimizes non-volatile residue contamination in vitreous ice. | Millipore Milli-Q or equivalent |
| Glow Discharge System (Argon/Oxygen) | Creates a hydrophilic, clean grid surface for even ice distribution and reduced contamination. | Pelco easiGlow, Quorum GloQube |
| Blotless Plunge Freezer | Eliminates filter paper contact, a major source of contaminants, ensuring pure, uniform ice. | SPT Labtech Chameleon |
| Volta Phase Plate | Enhances contrast in very thin ice, allowing use of thinner ice layers which are less prone to motion. | Thermo Fisher Scientific |
| Cryo-EM Grid Storage Box (Traceable) | Prevents frost accumulation and cross-contamination during grid storage in liquid nitrogen. | Scienion sciStorage, Tong |
| Advanced Buffer Additives (e.g., 0.01% Lauryl Maltose Neopentyl Glycol) | Stabilizes particles and reduces air-water interface interactions, a source of motion. | Anatrace Gold-Grade LMNG |
Resolving Symmetry Mismatches and Dealing with Flexible Regions (e.g., Glycans, Loops)
Application Notes
In the 3D reconstruction of virus particles from cryo-electron microscopy (cryo-EM) images, two major challenges impede high-resolution interpretation: symmetry mismatches and flexible regions. Viral capsids often exhibit pseudo-symmetry or local deviations from ideal symmetry, while surface glycoproteins and loop regions possess inherent flexibility. Overcoming these obstacles is critical for accurate structure-based drug and vaccine design.
1. Symmetry Mismatch Resolution: Many viruses, like Herpesviruses or Adenoviruses, incorporate portal proteins or other asymmetric complexes within an otherwise symmetric capsid. Enforcing perfect symmetry during reconstruction averages out these unique features. Current strategies involve localized reconstruction and symmetry relaxation. A key protocol is focused classification with signal subtraction, where the asymmetric region is masked and classified independently, allowing for its high-resolution determination without interference from the symmetric capsid.
2. Handling Flexible Regions: Viral surface proteins, such as the influenza hemagglutinin (HA) stalk or HIV-1 Env glycans, are highly dynamic. This flexibility leads to blurred density in reconstructions. Advanced image processing techniques, including 3D variability analysis and multi-body refinement, now allow for the explicit modeling of continuous conformational motions. For glycans, integrative modeling with mass spectrometry data and molecular dynamics simulations is becoming standard to define their heterogeneous conformations.
Quantitative Impact of Advanced Processing:
Table 1: Resolution Gains from Addressing Flexibility and Symmetry Mismatches
| Virus & Target | Standard Single-Particle Analysis (SPA) Resolution | After Multi-body/Focused 3D Classification Resolution | Key Technique Applied |
|---|---|---|---|
| Influenza A Virus (HA head domain) | 3.8 Å | 3.2 Å | Multi-body Refinement |
| HIV-1 Env Trimer (Glycan Shield) | 4.5 Å | 3.9 Å (core), Glycans defined | 3D Variability Analysis & AlphaFold2 integration |
| HSV-1 Capsid (Portal Vertex) | 4.0 Å (global) | 3.5 Å (portal complex) | Focused Classification & Local Refinement |
Experimental Protocols
Protocol 1: Focused Classification for Asymmetric Features Objective: To reconstruct a symmetry-mismatched viral portal complex.
- Initial Symmetric Reconstruction: Perform standard particle picking, 2D classification, and ab-initio 3D reconstruction in RELION or cryoSPARC, enforcing the icosahedral symmetry of the capsid.
- Signal Subtraction: Create a soft mask around the region of interest (e.g., a single vertex). Subtract all density outside this mask from each particle image, generating a new set of particle stacks containing only signal from the target region.
- 3D Classification: Perform 3D classification without symmetry (C1) on the subtracted particles. This will separate heterogeneous states of the portal complex (e.g., with/without DNA, different conformations).
- Local Refinement: Refine the selected class(es) from Step 3 using a high-resolution reference and a tight mask, still without imposing symmetry, to obtain the final asymmetric reconstruction.
Protocol 2: Multi-body Refinement for Flexible Glycoproteins Objective: To resolve independent motions of domains within a viral surface glycoprotein.
- Global Refinement: Generate a consensus reconstruction of the entire virus or spike protein at intermediate resolution (~4-5Å).
- Define Flexible Bodies: Using tools in RELION or ISOLDE/ChimeraX, define distinct rigid bodies (e.g., receptor-binding domain (RBD), glycan cap, fusion subunit). Each body must be a contiguous volume.
- Multi-body Analysis: Run multi-body refinement, which calculates optimal alignment for the entire particle while also determining the relative orientation and position of each defined body for every single particle image.
- Trajectory Analysis: The output includes a set of principal component trajectories showing the dominant motions. Generate 3D maps along these trajectories to visualize the continuum of states, from closed to open conformations.
Visualizations
Diagram 1: Focused Classification Workflow for Symmetry Mismatch
Diagram 2: Multi-body Refinement for Flexible Domains
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Advanced Virus 3D Reconstruction
| Item | Function & Application |
|---|---|
| Glycan-Restrictive Mutants | Engineered viruses with glycosylation sites knocked out to reduce heterogeneity and simplify density maps for core protein analysis. |
| Crosslinkers (e.g., GraFix, DSG) | Mild chemical crosslinking to stabilize transient conformations or virus-receptor complexes, reducing flexibility during grid preparation. |
| Fab Fragments / Nanobodies | High-affinity binders used to "lock" flexible glycoproteins (like HIV Env) into specific conformational states for classification. |
| Integrated Modeling Software (e.g., ISOLDE, Phenix) | Tools for real-time flexible fitting of atomic models into EM density, crucial for interpreting loop and glycan regions. |
| AlphaFold2 & RoseTTAFold | AI-predicted protein structures used as priors for modeling flexible regions, especially when density is weak or discontinuous. |
| High-Sensitivity Detectors (K3, G4) | Direct electron detectors enabling high-resolution data collection from small, fragile viruses and low-abundance conformational states. |
Application Notes: A Framework for High-Resolution Single-Particle Cryo-EM of Viral Structures
The pursuit of near-atomic resolution in the 3D reconstruction of virus particles via cryo-electron microscopy (cryo-EM) is a cornerstone of structural virology. It enables the precise mapping of capsid proteins, envelope glycoproteins, and drug-binding pockets. Achieving this requires meticulous optimization of data collection parameters and iterative refinement cycles. This protocol details a systematic approach to push resolution limits for icosahedral viruses, framed within a thesis on viral architecture and antiviral drug discovery.
Quantitative Parameter Optimization for Data Collection
High-resolution reconstruction begins at the microscope. The following parameters are interdependent and must be balanced.
Table 1: Key Data Collection Parameters and Optimized Ranges for Viral Particles
| Parameter | Typical Range (Optimized for Viruses) | Impact on Resolution & Data Quality |
|---|---|---|
| Defocus Range | -0.8 μm to -2.5 μm (staggered) | Provides CTF information; too little limits high-res, too much attenuates mid-res signal. |
| Electron Dose (Total) | 40-60 e⁻/Ų | Balances signal-to-noise and beam-induced motion damage. Higher dose damages specimen. |
| Dose Rate | 4-8 e⁻/pixel/sec (on camera) | Lower rates reduce coincidence loss and improve DQE of direct detectors. |
| Pixel Size (Physical) | 0.8 - 1.2 Å/pixel | Must satisfy Nyquist criterion for target resolution (e.g., 2.5 Å target needs ≤1.25 Å/pixel). |
| Number of Micrographs | 3,000 - 10,000+ | Depends on particle size, heterogeneity, and desired resolution. Viruses often require fewer. |
| Particles per Micrograph | 50 - 300+ | Maximizes data efficiency; depends on concentration and ice quality. |
| Movie Frames | 40-60 frames/movie | Enables optimal dose fractionation and motion correction. |
Protocol 1.1: Optimized Grid Screening and Data Acquisition
- Grid Preparation: Apply 3 μL of purified virus sample (≥0.5 mg/mL) to a freshly glow-discharged (30 sec, medium power) ultra-thin carbon or holey carbon grid (e.g., Quantifoil R 1.2/1.3). Blot for 3-5 seconds at 100% humidity, 4°C, and plunge-freeze in liquid ethane using a vitrification device (e.g., Vitrobot Mark IV).
- Microscope Setup: Load grid into a 300 keV cryo-TEM equipped with a post-column energy filter and a direct electron detector (e.g., Gatan K3 or Falcon 4). Operate the energy filter with a slit width of 20 eV.
- Initial Screening: At low magnification (e.g., 3,500x), assess ice quality and particle distribution. Target areas with ice thickness comparable to the virus diameter.
- Data Collection Setup:
- Switch to collection magnification (calibrated pixel size: 0.82 Å/pixel).
- Set a defocus range of -1.0, -1.5, and -2.0 μm in the acquisition software (e.g., SerialEM, EPU).
- Configure movie mode: 50 frames per movie, total dose 50 e⁻/Ų (1 e⁻/Ų/frame).
- Set the dose rate to 6 e⁻/pixel/sec on the detector.
- Enable coma-free beam-tilt and image-shift based multi-area acquisition.
- Automated Acquisition: Launch the session, collecting a minimum of 5,000 micrographs, targeting >200 particles per micrograph.
Iterative Refinement Cycle Strategy
Reconstruction is an iterative process of model improvement guided by resolution metrics (Fourier Shell Correlation, FSC).
Table 2: Key Metrics and Target Values for Refinement Cycles
| Metric | Calculation/Software | Target Value for High-Res |
|---|---|---|
| Global Resolution (FSC=0.143) | relion_postprocess or cryoSPARC 3D FSC |
< 3.0 Å |
| Map-Sharpening B-factor | relion_postprocess or phenix.auto_sharpen |
-50 to -150 Ų |
| Particle Alignment Accuracy | Angular accuracy reported by 3D refinement | < 1° |
| Per-Particle CTF Refinement | Defocus, astigmatism, beam tilt, per-micrograph B-factor | Improved particle FSC curves |
| Euler Angle Distribution | Histogram from refinement job | Even and complete coverage |
Protocol 2.1: High-Resolution Iterative Refinement Workflow
- Preprocessing: Motion-correct movies using
MotionCor2orRELION'simplementation. Estimate CTF parameters per micrograph usingCTFFIND-4.1orGctf. - Initial Model Generation: Pick particles (Template picker or
Topaz). Perform 2D classification to remove junk. Generate an ab initio model incryoSPARCor usingRELION3D initial model. - Cycle 1 - 3D Classification & Homogenization: Run heterogeneous refinement (
cryoSPARC) or 3D classification without alignment (RELION) with 3-4 classes to isolate intact, homogeneous viral particles. Select the best class(es). - Cycle 2 - High-Resolution Refinement: Refine the selected particles with a solvent mask, imposing icosahedral symmetry (I1). Perform CTF refinement to correct for defocus, astigmatism, and higher-order aberrations.
- Cycle 3 - Masked & Focused Refinement: Create a soft mask around a region of interest (e.g., a single capsid protein asymmetric unit). Run a focused 3D refinement without symmetry (C1) on this sub-region to reveal local flexibility at high resolution.
- Cycle 4 - Final Polishing & Validation: Apply Bayesian polishing (
RELION) or particle-based motion correction. Perform a final non-uniform refinement (cryoSPARC). Validate the final map usingphenix.mtriageandEMRingerscore. Calculate the FSC between the final map and an atomic model inphenix.validation_cryoem.
Diagram 1: High-Res Cryo-EM Workflow for Viruses
Diagram 2: The Iterative Refinement Feedback Loop
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Viral Cryo-EM
| Item | Function & Rationale |
|---|---|
| Ultra-Thin Carbon Films on Holey Carbon Grids (e.g., Quantifoil R 1.2/1.3) | Provides a stable, thin support film over holes for particles to be suspended in vitreous ice, minimizing background noise. |
| Liquid Ethane (Research Grade) | Cryogen with high heat capacity for rapid vitrification, preventing crystalline ice formation that destroys specimen high-resolution detail. |
| Glow Discharger | Creates a hydrophilic surface on grids, ensuring even sample spread and thin ice. Critical for reproducibility. |
| Holey Gold Grids (e.g., UltraAuFoil) | Gold is more conductive than copper, reducing charging and drift during imaging, especially for large viruses. |
| Size-Exclusion Chromatography (SEC) Buffer (e.g., Tris-HCl, NaCl, pH 7.5) | Final purification step to isolate monodisperse, intact virus particles and exchange into a clean, volatile-free buffer. |
| Tannic Acid / GraFix (Gradient Fixation) | Chemical stabilizer used sparingly for fragile complexes to improve particle integrity, though may limit ultimate resolution. |
| Ammonium Molybdate (2% w/v, negative stain) | For rapid initial negative stain EM to assess sample quality, concentration, and monodispersity before committing to cryo-EM. |
| Direct Electron Detector (e.g., Gatan K3, Falcon 4) | Camera with high detective quantum efficiency (DQE) and fast frame rates, enabling dose fractionation; the single most impactful hardware for resolution. |
Ensuring Fidelity: Validation Metrics and Comparative Structural Biology
In the broader thesis on 3D reconstruction of virus particles from cryo-electron microscopy (cryo-EM) images, quantitative validation is the cornerstone for assessing map reliability. For structural virology and rational drug design, the resolution and fidelity of a reconstructed virus capsid or glycoprotein complex directly impact the interpretation of epitopes, binding sites, and conformational states. Fourier Shell Correlation (FSC) serves as the primary metric for global resolution estimation, while Map-Model FSC validates atomic model fitting against the map. Local resolution analysis reveals heterogeneity, crucial for understanding asymmetric features or flexible regions in viral particles.
Core Concepts & Quantitative Data
Fourier Shell Correlation (FSC)
FSC measures the normalized cross-correlation coefficient between two independent 3D reconstructions (e.g., from half-maps) across successive spherical shells in Fourier space. The resolution at the FSC=0.143 threshold (gold-standard) is the most widely accepted criterion.
Table 1: Common FSC Thresholds and Interpretations
| Threshold Value | Common Name | Interpretation in Virus Reconstruction |
|---|---|---|
| FSC = 0.5 | FSC 0.5 | Historical threshold; often overestimates resolution. |
| FSC = 0.143 | Gold Standard | Based on σ=1 criterion; standard for map-to-map resolution. |
| FSC = 0.0 | – | Theoretical point where noise dominates. |
Map-Model FSC
This validation metric correlates the map computed from an atomic model (e.g., of a viral capsid protein) with the experimental cryo-EM map. It assesses model accuracy.
Table 2: Map-Model FSC Interpretation Guidelines
| FSC Range (Mean) | Model-to-Map Fit Assessment | Implication for Drug Design |
|---|---|---|
| > 0.8 | Excellent fit | High-confidence structure for docking. |
| 0.6 - 0.8 | Good fit | Reliable for identifying binding pockets. |
| 0.5 - 0.6 | Moderate fit | Caution advised; model may need refinement. |
| < 0.5 | Poor fit | Model not well supported by the map. |
Local Resolution
Variations in resolution across a 3D map, calculated using methods like blocres or ResMap, highlight regions of stability or flexibility.
Table 3: Local Resolution Ranges in a Typical Virus Reconstruction
| Resolution Range (Å) | Structural Features Typically Resolved | Example in Virology |
|---|---|---|
| 2.0 - 3.0 | Side chains, water networks | Ordered core of capsid protein. |
| 3.0 - 4.5 | Polypeptide backbone, large side chains | Majority of the capsid shell. |
| 4.5 - 6.0 | Secondary structure elements (α-helices, β-sheets) | Flexible linkers between domains. |
| > 6.0 | Overall molecular shape | Highly mobile glycans or termini. |
Experimental Protocols
Protocol: Calculating Gold-Standard FSC for a Virus Reconstruction
Purpose: To determine the global resolution of a cryo-EM map from two independently refined half-maps. Software: RELION, cryoSPARC, or EMAN2. Steps:
- Particle Splitting: During 3D auto-refinement, randomly assign your extracted virus particle images into two independent sets (Half-set A and Half-set B).
- Independent Reconstruction: Reconstruct two 3D maps (half-maps) from each set, using identical parameters but separate Fourier space information.
- Mask Application: Apply a soft-edged mask that tightly encloses the viral particle to exclude noise from the solvent. This is critical for accurate FSC.
- Fourier Transformation: Compute the 3D Fourier transform of each masked half-map.
- Shell Correlation: For each spherical shell in Fourier space (defined by spatial frequency, s), calculate: FSC(s) = Σ [F1(s) • F2(s)] / sqrt[ Σ|F1(s)|² • Σ|F2(s)|² ]* where F1 and F2 are Fourier coefficients, and * denotes complex conjugate.
- Plotting & Thresholding: Plot FSC(s) vs. spatial frequency (1/Å). The resolution is reported as the spatial frequency where the FSC curve crosses the 0.143 threshold. Interpolate if necessary.
Protocol: Computing Map-Model FSC
Purpose: To validate an atomic model (e.g., PDB file) against the experimental cryo-EM map. Software: PHENIX (phenix.mtriage, phenix.modelvsmap), CCP-EM, or UCSF ChimeraX. Steps:
- Model Preparation: Ensure your atomic model (e.g., of a viral capsid) is properly positioned (fitted) into the experimental map.
- Map Calculation from Model: Using a program like
phenix.molmaporpdb2map, compute a simulated map from the atomic model at the same grid spacing (pixel size) as your experimental map. Apply a B-factor to approximate the fall-off of high-resolution information. - Masking: Apply a common mask (around the model) to both the experimental and the simulated model maps.
- FSC Calculation: Perform an FSC calculation (as in 3.1, but now between the experimental map and the simulated model map).
- Analysis: Report the FSC curve and the mean FSC value (typically over a specified resolution range, e.g., 10.0Å to the reported map resolution). A mean FSC > 0.8 indicates good model-to-map agreement.
Protocol: Estimating Local Resolution with Blocres
Purpose: To generate a local resolution map highlighting regions of varying clarity.
Software: Bsoft (blocres), cryoSPARC, or ResMap.
Steps:
- Input Half-maps: Provide the two unfiltered, masked half-maps from the gold-standard reconstruction.
- Window Definition:
blocresdivides the 3D map into small, overlapping sub-volumes (blocks). A typical block size is 10-20 voxels, smaller than the viral particle but large enough for FSC calculation. - Local FSC per Block: For each block, a local FSC curve is calculated between the two half-maps.
- Thresholding: The spatial frequency at which the local FSC drops below 0.5 (or sometimes 0.143) is assigned as the local resolution for the central voxel of that block.
- Interpolation & Output: Values are interpolated across blocks to create a continuous 3D local resolution map, which can be color-coded and overlaid on the density map for visualization.
Visualizations
Title: Gold-Standard FSC Calculation Workflow
Title: Relationship Between Validation Metrics and Drug Design
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials & Software for Quantitative Validation in Cryo-EM Virology
| Item Name | Category | Function in Virus Particle Reconstruction |
|---|---|---|
| RELION 4.0 | Software | Performs gold-standard 3D refinement, FSC calculation, and post-processing including masking. |
| cryoSPARC v4 | Software | Offers live, per-iteration FSC monitoring, local resolution estimation (cryoSPARC-LocalRes), and non-uniform refinement. |
| PHENIX Suite | Software | Provides comprehensive tools for Map-Model FSC (phenix.mtriage), model refinement, and validation. |
| UCSF ChimeraX | Software | Visualizes local resolution maps as color overlay on density and calculates map-model correlation. |
| EMDB Map Mask | Data/Utility | A soft-edged, spherical mask tailored for icosahedral virus particles to isolate capsid signal from solvent for FSC. |
| High-Performance GPU Cluster | Hardware | Accelerates iterative refinement and FSC calculations, essential for large virus particle datasets (>100k particles). |
| B-factor Plot | Analytical Tool | Graph of per-shell B-factor from post-processing; indicates resolution fall-off and guides map sharpening. |
Within the broader thesis on 3D reconstruction of virus particles from cryo-electron microscopy (cryo-EM) images, rigorous validation is paramount. The low signal-to-noise ratio inherent to cryo-EM, coupled with complex iterative refinement algorithms, creates significant risks of overfitting and model bias. This document details application notes and protocols for employing cross-validation techniques—specifically the gold-standard Fourier Shell Correlation (FSC), overfitting detection methodologies, and understanding masking effects—to ensure the biological fidelity of reconstructed viral structures for downstream research and drug development.
The Gold-Standard FSC Protocol
Core Concept & Quantitative Benchmarks
The gold-standard FSC procedure splits the experimental dataset into two independent halves during the entire refinement and reconstruction process. The final 3D maps from each half-set are compared in Fourier space to calculate a resolution estimate. This prevents information leakage and provides an unbiased validation metric.
Table 1: FSC Thresholds and Interpretation
| FSC Value | Common Threshold Name | Interpretation in 3D EM |
|---|---|---|
| 0.143 | "Gold-Standard" Resolution | Conservative estimate; corresponds to (\sim)1/2 bit information per voxel. Industry standard for published maps. |
| 0.5 | "FSC(_{0.5})" | Traditional threshold; considered over-optimistic without gold-standard. |
| 0.0 | Nominal Correlation | Point where no significant signal remains. Used with caution. |
| FSC between half-maps vs FSC of half-map to full map | Overfitting Detection | A significant gap ((>)~0.05-0.1) at high resolution indicates overfitting. |
Experimental Protocol: Gold-Standard Refinement & FSC Calculation
Materials: Particle stack (e.g., 100,000 virus particle images), refinement software (RELION, cryoSPARC, etc.), high-performance computing cluster.
Procedure:
- Initial Model Generation: Generate an ab initio model from a small, randomly selected subset of particles (e.g., 10%) using stochastic gradient descent (cryoSPARC) or class averages. This model must not be used as a reference for the subsequent refinement of the half-sets.
- Random Split: Randomly divide the entire particle dataset into two independent halves (Half-set A and Half-set B). Ensure splits are performed on the particle image level, not the micrograph level, to mitigate pseudo-independence.
- Independent Refinement: Refine Half-set A and Half-set B completely independently starting from the same initial model (Step 1). This includes all iterations of alignment, classification (if performed), and reconstruction. Use identical refinement parameters for both halves.
- Final Map Reconstruction: Generate the final post-processed map for each half-set. Apply any masking or filtering after the FSC calculation for resolution estimation, or note carefully if applied before.
- FSC Calculation: a. Compute the 3D Fourier transform of each half-map. b. For each spherical shell in Fourier space (defined by spatial frequency s), calculate the correlation coefficient between the Fourier components of the two half-maps. [ \text{FSC}(s) = \frac{\sum{\vec{s} \in \text{shell}} F1(\vec{s}) \cdot F2^*(\vec{s})}{\sqrt{\sum |F1(\vec{s})|^2 \cdot \sum |F_2(\vec{s})|^2}} ] c. Plot FSC(s) against spatial frequency (1/Å).
- Resolution Determination: Report the spatial frequency where the FSC curve crosses the 0.143 threshold. This is the reported gold-standard resolution of the map.
Diagram Title: Gold-Standard FSC Workflow
Overfitting Detection Protocols
Core Concept
Overfitting occurs when the refinement algorithm models noise or reconstruction artifacts specific to the dataset rather than the true biological signal. The gold-standard FSC is the primary defense, but additional checks are necessary.
Table 2: Overfitting Diagnostics
| Diagnostic Test | Procedure | Interpretation of Overfitting |
|---|---|---|
| FSC({work}) vs FSC({test}) | Refine half-maps (work), compare each to the map from the other half (test). | A large gap ((>)0.1) between the work and test curves at mid-to-high resolution. |
| Model vs Map FSC | Calculate FSC between the final refined atomic model and the map it was refined against. | The FSC curve peaks significantly below 1.0 at low resolution, indicating the map lacks features present in the model. |
| Randomization (Shuffling) Test | Apply random phases to particle images beyond a certain resolution and re-refine. | The refined map recovers spurious high-resolution features from randomized data. |
| Tilt-Pair Validation | Use particle pairs from tilted micrographs to validate particle orientations. | Reported orientations do not predict the second view of the tilt pair accurately. |
Experimental Protocol: FSC({work}) vs FSC({test}) Analysis
This protocol runs concurrently with Section 1.2.
Procedure:
- After independent refinement (Step 1.2.3), generate three maps: Half-map A, Half-map B, and a combined map (from all particles, refined against the combined set after independent half-set refinements have finalized orientations and classes).
- Calculate FSC(_{work}): Correlation between Half-map A and Half-map B (this is the standard gold-standard FSC).
- Calculate FSC(_{test}): For each half-set, calculate the FSC between that half-map and the other half-set's reconstruction (e.g., Half-map A vs. the map from Half-set B). In practice, this is often approximated by calculating the FSC between each half-map and the combined map.
- Plot and Analyze: Plot both FSC({work}) and FSC({test}) on the same graph.
- Diagnosis: A close alignment of the two curves indicates minimal overfitting. A significant separation, where FSC({work}) is substantially higher than FSC({test}) at intermediate-to-high frequencies, indicates the model has overfit to noise in each work set.
Diagram Title: Overfitting Detection Logic
Understanding and Controlling Masking Effects
Core Concept: The Masking Paradox
Applying a soft mask to the 3D map during post-processing improves the FSC resolution by isolating the particle from surrounding solvent noise. However, an overly tight or aggressive mask can artificially inflate the FSC by correlating noise inside the mask boundary, a phenomenon known as the "masking effect" or "Einstein from noise."
Table 3: Masking Effects on FSC
| Mask Type | Effect on Gold-Standard FSC | Risk | Recommended Practice |
|---|---|---|---|
| No Mask (Spherical Shell) | Unbiased but low resolution due to solvent noise. | Under-estimation of true resolution. | Use as a baseline. |
| Loose Mask (Dilated from threshold) | Moderate improvement. Lower risk of inflation. | May include too much solvent noise. | Good starting point for final resolution. |
| Tight Mask (Eroded from threshold) | High apparent resolution. | High risk of artificial inflation, overfitting. | Avoid for primary resolution report. Use with extreme caution. |
| Composite Mask (e.g., Solvent + Protein) | Can be beneficial for multi-component structures. | Complexity can hide bias. | Validate with independent methods (model-based). |
Experimental Protocol: Safe Mask Application for Validation
Materials: Unmasked half-maps, masking software (RELION relion_mask_create, UCSF Chimera, etc.).
Procedure:
- Generate a Conservative Mask: From the combined (unmasked) map, threshold at a level that clearly separates particle density from solvent. Use automated tools (e.g.,
relion_mask_create --ini_threshold 0.02). Dilate this binary mask by 5-10 pixels to ensure it encompasses all potential signal and the "soft edge" region. - Apply a Soft Edge: Apply a soft cosine edge (e.g., 5-10 pixels wide) to the dilated mask to prevent Fourier artifacts.
- Apply Mask for Post-Processing: Apply this same soft mask to both unmasked half-maps before the final FSC calculation for resolution estimation.
- Sensitivity Analysis: Repeat the FSC calculation using masks generated with slightly higher and lower initial thresholds and dilation values.
- Report Transparently: Always report the mask volume, dilation, and soft edge parameters alongside the resolution. The stated resolution is only valid for the masked region.
Diagram Title: Safe Mask Generation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for Cryo-EM Validation Experiments
| Item / Reagent | Function in Validation Protocol | Example / Notes |
|---|---|---|
| Cryo-EM Grids (Au, 300 mesh) | Support for vitrified virus sample. | Quantifoil R1.2/1.3, UltrAuFoil. |
| Vitrification System | Creates amorphous ice embedding virus particles. | Thermo Fisher Vitrobot Mark IV, Leica EM GP2. |
| 300 kV Cryo-Electron Microscope | High-resolution data acquisition. | Thermo Fisher Krios, Talos Arctica, JEOL CRYO ARM. |
| Direct Electron Detector | Captures high-fidelity, dose-fractionated movies. | Gatan K3, Falcon 4, Selectris X. |
| Processing Software Suite | Particle picking, 2D/3D classification, refinement, and FSC calculation. | RELION, cryoSPARC, EMAN2, Scipion. |
| High-Performance Computing Cluster | Enables computationally intensive gold-standard refinements. | CPU/GPU nodes with >1TB RAM and high-speed storage. |
| Visualization & Modeling Software | Map analysis, masking, model building, and validation. | UCSF Chimera/X, Coot, Phenix, ISOLDE. |
| Validation Servers & Packages | Independent assessment of map and model quality. | EMDB validation server, PHENIX comprehensive validation, MolProbity. |
Within the context of a broader thesis on the 3D reconstruction of virus particles from electron microscopy (EM) images, understanding the complementary roles of structural biology techniques is paramount. This document provides detailed Application Notes and Protocols for utilizing Cryo-Electron Microscopy (Cryo-EM), X-ray Crystallography, and Nuclear Magnetic Resonance (NMR) spectroscopy. Each technique offers unique strengths for elucidating viral architecture, informing drug design, and understanding pathogenesis. The synergy between these methods is increasingly crucial for tackling complex virological challenges.
Quantitative Comparison of Core Techniques
Table 1: Key Quantitative Metrics for Structural Biology Techniques (as of 2024)
| Parameter | Single-Particle Cryo-EM | X-ray Crystallography | Solution NMR | Solid-State NMR |
|---|---|---|---|---|
| Typical Size Range | >50 kDa – Giant complexes (MDa), intact virions | <10 kDa – Large complexes (>1 MDa) * | <50 kDa (in solution) | No strict upper limit (membranes, assemblies) |
| Specimen State | Vitrified solution, near-native | Crystalline solid | Solution, native conditions | Solid/amorphous powder, oriented membranes |
| Typical Resolution | 1.8 – 4.0 Å (routine); sub-Å possible | 0.8 – 3.0 Å (atomic) | 1 – 3 Å (backbone); 3 – 6 Å (side chains) | 1 – 4 Å (local); 10 – 20 Å (global) |
| Sample Requirement (Conc.) | ~0.05–1 mg/mL (3–5 µL) | 5–20 mg/mL (10s of nL – µL) | 0.3–1 mM (300–500 µL) | ~5–20 mg (rotor) |
| Data Collection Time | Hours to days (direct detector) | Minutes to hours (synchrotron) | Days to weeks | Days to weeks |
| Temperature | ~-180°C (cryogenic) | 100K (cryo) or room temp | 4–40°C | ~-150 to 50°C |
| Key Output | 3D density map, atomic models, conformational states | Atomic coordinates, B-factors (disorder) | Atomic coordinates, dynamics (ps-ns), interactions | Atomic constraints, dynamics (ms-s), orientation |
| Primary Virus Application | Intact virions, asymmetric complexes, conformational heterogeneity | Atomic details of purified viral proteins/domains | Dynamics of small viral proteins, unfolded regions, drug interactions | Capsid assemblies, membrane-bound viral proteins, enveloped viruses |
*Requires crystallization, which is size and complex-dependent.
Table 2: Suitability for Key Virology Research Questions
| Research Goal | Optimal Technique(s) | Rationale & Synergy |
|---|---|---|
| High-Resolution Atomic Model of Stable Viral Protein | X-ray Crystallography | Unmatched precision for well-ordered, crystallizable targets (e.g., protease active site). |
| Structure of Intact, Asymmetric Virion | Single-Particle Cryo-EM | Handles large, non-crystalline particles (e.g., ~1000 Å Herpesvirion). No crystal packing artifacts. |
| Conformational Dynamics of Viral Fusion Protein | Cryo-EM + NMR/MD | Cryo-EM: snapshots of states. NMR/MD: atomic-level dynamics and energy landscapes. |
| Drug Binding to Flexible Viral RNA Element | NMR + X-ray/Cryo-EM | NMR identifies binding site/dynamics in solution. X-ray/Cryo-EM provides high-res context in complex. |
| Membrane-Bound Viral Channel Protein | Solid-State NMR + Cryo-ET | ssNMR gives atomic detail in lipid environment. Cryo-ET situates it in cellular context. |
| Rapid Screening of Antiviral Compounds | X-ray Fragment Screening + Cryo-EM Validation | X-ray rapidly IDs fragment hits. Cryo-EM validates binding in native-like complex. |
Detailed Experimental Protocols
Protocol 1: Single-Particle Cryo-EM for Intact Virus Particle Reconstruction
Context: Determining the architecture of a ~500 Å non-enveloped icosahedral virus.
A. Virus Purification & Grid Preparation
- Purify virus from cell culture via ultracentrifugation (sucrose gradient) to >95% homogeneity.
- Quantify concentration to ~5–10 mg/mL using UV spectrophotometry (A260/A280).
- Apply 3 µL of sample to a glow-discharged (15–30 sec, air plasma) Quantifoil R 1.2/1.3 Au 300 mesh grid.
- Blot for 3–5 seconds at 100% humidity, 4°C (Vitrobot Mark IV), and plunge-freeze into liquid ethane.
B. Data Collection on a 300 keV Cryo-TEM
- Screen grids for ice quality and particle distribution at low magnification (80,000x).
- Collect micrographs in super-resolution mode on a K3 direct electron detector.
- Defocus range: -0.8 to -2.5 µm.
- Total dose: ~50 e-/Ų, fractionated over 40 frames.
- Nominal magnification: 81,000x (0.55 Å/pixel super-res).
- Use beam-image shift to collect 200-500 images/grid hole.
C. Image Processing & 3D Reconstruction (Relion/CryoSPARC Workflow)
- Pre-process: Motion correction (MotionCor2), CTF estimation (CTFFIND-4.1).
- Particle picking: Template-based (from 2D classes) or AI-based (Topaz/cryoSPARC Live).
- 2D Classification: Extract ~1,000,000 particles, classify to remove junk/ice.
- Ab-initio & Heterogeneous Refinement: Generate initial model de novo, sort particle heterogeneity.
- High-Resolution 3D Refinement: For homogeneous subset, refine imposing icosahedral symmetry (I1).
- Bayesian Polishing & CTF Refinement: Correct per-particle beam-induced motion and aberrations.
- Final Reconstruction: Iterate refinement until convergence. Use gold-standard FSC to determine resolution (e.g., 2.8 Å at FSC=0.143).
D. Model Building & Validation
- Fit available high-resolution X-ray structures of capsid proteins into Cryo-EM density (ChimeraX).
- Build de novo models for unresolved regions using Coot, guided by sequence.
- Real-space refine model against map using Phenix or ISOLDE.
- Validate using MolProbity (clashscore, rotamers) and map-model FSC.
Protocol 2: X-ray Crystallography of a Viral Protease with Inhibitor
Context: Obtaining atomic detail of drug binding for structure-based optimization.
A. Protein Expression, Purification & Crystallization
- Express recombinant viral protease in E. coli with a cleavable His-tag.
- Purify via Ni-NTA affinity, tag cleavage, and size-exclusion chromatography (Superdex 75).
- Concentrate to 10 mg/mL in 20 mM HEPES pH 7.5, 150 mM NaCl.
- Set up 96-well sitting-drop vapor diffusion plates: Mix 100 nL protein + 100 nL reservoir.
- Screen commercial sparse-matrix screens (e.g., Morpheus, JC SG). Incubate at 20°C.
- Optimize hit condition (pH, precipitant, additive) to grow large (>50 µm), diffraction-quality crystals.
B. Soaking & Data Collection
- Transfer a single crystal to reservoir solution containing 5 mM inhibitor (from 100 mM DMSO stock).
- Soak for 1–2 hours.
- Cryo-protect by transferring to reservoir + 25% ethylene glycol. Flash-cool in liquid N2.
- Collect 360° of data at 100K on a synchrotron beamline (e.g., Diamond I24, microfocus) with a DECTRIS EIGER2 XE detector. Exposure 0.1 sec/deg, flux attenuated for completeness >99%.
C. Structure Solution & Refinement
- Process data: Index, integrate, scale (XDS/AIMLESS). Space group P2₁2₁2₁.
- Solve by molecular replacement (Phaser) using apo protease structure as search model.
- Build model: Refine cycles (phenix.refine) with manual rebuilding (Coot). Add inhibitor to clear Fo-Fc density.
- Finalize: Add waters, DMSO. Validate with Rwork/Rfree (~18%/22%) and MolProbity.
Protocol 3: NMR Analysis of Viral Protein Dynamics & Ligand Interaction
Context: Characterizing the dynamics of a disordered N-terminal domain of a capsid protein.
A. Isotope Labeling & Sample Preparation
- Express protein in M9 minimal media with [15N]NH4Cl and/or [13C]glucose as sole nitrogen/carbon sources.
- Purify as in Protocol 2A. Dialyze into NMR buffer (20 mM phosphate pH 6.5, 50 mM NaCl, 0.5 mM TCEP, 10% D2O).
- Concentrate to 0.3 mM in 280 µL (Shigemi tube).
B. NMR Data Acquisition (800 MHz Spectrometer)
- Collect 2D [1H-15N] HSQC at 25°C (standard Bruker pulse sequence, topspin).
- For assignment: Acquire 3D HNCACB, CBCA(CO)NH, HNCO, HN(CA)CO.
- For dynamics: Acquire [15N] T1, T2 relaxation series and {1H}-15N heteronuclear NOE.
- For titration: Record a series of 2D [1H-15N] HSQCs upon addition of small molecule ligand (0.1 to 2 molar eq).
C. Data Processing & Analysis
- Process data (NMRPipe). Fourier transform, phase, baseline correct.
- Assign backbone nuclei (CARAVEL/CCPNMR Analysis). Map peaks to sequence.
- Analyze Chemical Shift Perturbation (CSP): Δδ = √((ΔδH)² + (ΔδN/5)²). Residues with Δδ > mean + 1σ indicate binding site.
- Analyze Relaxation Data: Fit T1/T2 to extract rotational correlation time. Low NOE values indicate ps-ns timescale flexibility.
Visualizations
Title: Cryo-EM 3D Reconstruction Workflow
Title: Synergistic Integration of Structural Techniques
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Structural Virology
| Category | Item | Function & Application | Example Vendor/Product |
|---|---|---|---|
| Sample Prep (Cryo-EM) | Quantifoil/C-flat Holey Carbon Grids | Support film with holes for vitrified sample suspension. Au grids preferred for viruses. | Quantifoil R 1.2/1.3 Au 300 mesh |
| Vitrification Robot | Standardizes plunge-freezing for reproducible, vitreous ice. | Thermo Fisher Vitrobot Mark IV | |
| Sample Prep (X-ray) | Crystallization Sparse-Matrix Screens | Empirically screen chemical space for crystallization conditions. | Molecular Dimensions Morpheus JC SG |
| Microfocus Synchrotron Beamline | Enables data collection from micro-crystals and radiation-sensitive samples. | Diamond Light Source I24 | |
| Data Collection | Direct Electron Detector (DED) | Captures high-resolution images with high DOE at low dose for Cryo-EM. | Gatan K3, Falcon 4 |
| High-Field NMR Spectrometer | Provides sensitivity and resolution for biomolecular NMR, including large complexes. | Bruker NEO 800/900 MHz | |
| Software & Analysis | Cryo-EM Processing Suite | Integrated software for processing, reconstruction, and analysis. | cryoSPARC Live, RELION-4 |
| Molecular Graphics & Modeling | Fitting, building, and refining atomic models into Cryo-EM/X-ray density. | UCSF ChimeraX, Coot | |
| NMR Data Processing & Analysis | Suite for processing, assigning, and analyzing multidimensional NMR data. | NMRPipe, CARAVEL | |
| Specialized Reagents | Deuterated/Isotope-Labeled Media | Produces 13C/15N-labeled proteins for NMR spectroscopy and structural studies. | Silantes D-M9 kits |
| Lipid Nanodiscs/KiNets | Membrane mimetics for studying membrane proteins in Cryo-EM/NMR. | MSP Nanodiscs (Cube Biotech) |
The primary thesis in modern structural virology posits that a complete understanding of viral assembly, infection, and neutralization requires integration of atomic-resolution 3D architectures with dynamic compositional and interaction data. While single-particle cryo-electron microscopy (cryo-EM) provides high-resolution structural maps of viral particles, it often lacks definitive identification of protein components, post-translational modifications (PTMs), and transient host-factor interactions. This Application Note details protocols for synergizing cryo-EM with mass spectrometry (MS) and bioinformatics to create comprehensive models of virus particles, crucial for rational vaccine and antiviral drug design.
Application Notes: Synergistic Data Integration
Table 1: Multi-Scale Data Types and Resolutions for Virus Particle Analysis
| Data Type | Typical Resolution/Accuracy | Information Gained | Key Limitation Addressed by Integration |
|---|---|---|---|
| Cryo-EM (Single-Particle) | 2.0 – 3.5 Å (Local), 3-8 Å (Whole Particle) | 3D Density map, capsid symmetry, glycoprotein spikes | Component identification, stoichiometry |
| Native Mass Spectrometry (nMS) | ~0.01% mass accuracy | Mass of intact complexes, stoichiometry, oligomeric states | Assigning density to specific proteins |
| Cross-linking MS (XL-MS) | 10-30 Å residue pair distance | Proximal residues, protein-protein interfaces, topology | Validating and refining low-resolution regions |
| Bottom-up Proteomics | Peptide sequence identity | Protein composition, PTMs (glycosylation, phosphorylation), sequence variants | Identifying modified residues in density |
| Bioinformatics (Sequence Analysis) | N/A | Conservation, domain prediction, disorder, epitope mapping | Functional annotation of structural features |
Table 2: Published Examples of Integrated Studies (2022-2024)
| Virus Studied | Cryo-EM Resolution | Integrated MS/Bioinformatics Method | Key Discovery | Reference (Type) |
|---|---|---|---|---|
| SARS-CoV-2 Omicron Spike | 2.8 Å | Glycoproteomics & MD Simulations | Defined glycan shield remodeling | Preprint 2023 |
| HIV-1 Envelope Trimer | 3.2 Å | Native MS & HDX-MS | Stabilizing mutation effects on conformation | Nature Comm. 2023 |
| Herpesvirus Capsid | 4.5 Å (asymmetric) | Cross-linking MS (XL-MS) | Tegument protein attachment sites | Science Adv. 2022 |
| Influenza A Virus | 3.7 Å (whole virion) | Lipidomics & Ion Mobility MS | Host lipid composition and membrane curvature | Cell 2024 |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Integrated Virus Particle Analysis
| Item | Function in Protocol | Example Product/Catalog # | Critical Notes |
|---|---|---|---|
| Grids for Cryo-EM | Support film for vitrified samples | Quantifoil R1.2/1.3 Au 300 mesh | Holey carbon film for optimal ice thickness. |
| GraFix Sucrose/Glycerol Gradient Kit | Stabilize complexes for nMS & cryo-EM | Separates intact virions from aggregates. | |
| BS³ (bis(sulfosuccinimidyl)suberate) | Water-soluble amine-reactive crosslinker for XL-MS | Thermo Fisher #21580 | Compatible with native conditions. |
| TFE (Trifluoroethanol) | For on-grid digestion in EM-MS | Enables in-situ proteolysis for MS analysis. | |
| Ammonium Acetate (≥250 mM) | Volatile buffer for native MS | Replaces non-volatile salts during buffer exchange. | |
| PNGase F | Enzyme for deglycosylation in glycoproteomics | Removes N-linked glycans to simplify MS spectra. | |
| Cryo-EM Map Sharpening Tool | Software for enhancing map interpretability | DeepEMhancer, Phenix.auto_sharpen | Aids in fitting MS-identified components. |
| Integrative Modeling Platform (IMP) | Software for combining all data types | Generates consensus structural models. |
Detailed Experimental Protocols
Protocol A: Concurrent Sample Preparation for Cryo-EM and Native MS
Objective: To prepare a homogeneous sample of purified virus particles suitable for both high-resolution cryo-EM and native MS analysis.
Materials: Purified virus suspension, Amicon Ultra centrifugal filters (100kDa MWCO), ammonium acetate solution (250 mM, pH 7.5), GraFix gradient components.
Procedure:
- Concentration & Buffer Exchange: Concentrate 200 µL of purified virus (~0.5 mg/mL) using a 100kDa MWCO centrifugal filter at 4,000 x g, 4°C. Exchange into 250 mM ammonium acetate (pH 7.5) via three cycles of dilution and concentration.
- Sample Split: Divide the concentrated sample into two aliquots (Aliquot C for Cryo-EM, Aliquot M for MS).
- Aliquot C (Cryo-EM): Dilute to ~3 µM concentration. Apply 3 µL to a freshly glow-discharged Quantifoil grid. Blot for 3-4 seconds and plunge-freeze in liquid ethane using a Vitrobot (100% humidity, 4°C).
- Aliquot M (Native MS): Further concentrate to ~10 µM. Load into a gold-coated nano-ESI capillary. Acquire spectra on a Q-TOF instrument equipped for high-mass detection (capillary voltage 1.2-1.6 kV, pressure 5-7 mbar).
Protocol B: Cross-linking Mass Spectrometry (XL-MS) on Grid
Objective: To capture transient interactions and protein interfaces within vitrified virus particles directly on the EM grid.
Materials: BS³ crosslinker (fresh 20 mM stock in H2O), 0.5M Ammonium Bicarbonate, Quenching solution (1M Tris-HCl, pH 7.5).
Procedure:
- On-Grid Cross-linking: Apply 3 µL of BS³ solution (2 mM final) to a blotted (but not vitrified) EM grid for 30 seconds at room temperature.
- Quenching: Wick away crosslinker and immediately apply 3 µL of 100 mM Tris-HCl (pH 7.5) for 30 seconds to quench the reaction.
- Vitrification: Blot and plunge-freeze as in Protocol A.
- In-Gel Digestion: Thaw the grid, wash with ammonium bicarbonate. Transfer the sample from the grid to a tube. Reduce (DTT), alkylate (IAA), and digest overnight with trypsin/Lys-C.
- LC-MS/MS Analysis: Analyze peptides on a Orbitrap Eclipse using a 60-min gradient. Use search software (e.g., pLink2, XlinkX) to identify cross-linked peptide pairs.
Protocol C: Integrative Modeling Workflow using Cryo-EM, MS, and Bioinformatics
Objective: To generate a consensus 3D model of a virus particle by fitting and refining atomic coordinates against all available data.
Procedure:
- Data Input: Collect (a) Cryo-EM density map (
.mrc), (b) XL-MS distance restraints (residue pairs < 30 Å), (c) Native MS mass constraints for subcomplexes, (d) Protein sequences (.fasta). - De Novo Modeling: Use Alphafold2 or RoseTTAFold to generate initial models for unknown protein components.
- Density Fitting: Rigid-body fit all component models into the cryo-EM map using UCSF ChimeraX.
- Restraint-Driven Refinement: In the Integrative Modeling Platform (IMP), apply spatial restraints from XL-MS and mass data. Run Monte Carlo sampling to satisfy all restraints simultaneously.
- Model Validation: Calculate a cross-correlation coefficient between the final model map and the cryo-EM map. Validate XL-MS satisfaction (percentage of cross-links within the maximum allowed distance).
Visualization Diagrams
Multi-Scale Data Integration Workflow for Virus Reconstruction
Integrative Model Refinement Cycle
The field of 3D reconstruction of virus particles from electron microscopy (EM) images has been revolutionized by the establishment of public, structured data repositories. The Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB) serve as the foundational pillars for archiving, validating, and disseminating structural data. Within the thesis context of advancing virus particle reconstruction, deposition into these repositories is not merely an endpoint but a critical step for validation, collaboration, and accelerating drug discovery. These resources ensure reproducibility, facilitate the development of new computational methods, and provide essential data for structure-based vaccine and antiviral drug design.
Table 1: Core Features of EMDB and PDB
| Feature | EMDB (Electron Microscopy Data Bank) | PDB (Protein Data Bank) |
|---|---|---|
| Primary Content | 3D EM maps & associated metadata, tomography data. | Atomic coordinates of proteins, nucleic acids, complexes (from X-ray, NMR, EM). |
| Virus-Specific Entries (approx.) | >6,500 maps (as of 2024) | >12,000 virus-related structures (as of 2024) |
| Key Deposition Mandates | Final map, map metadata (resolution, method), sample prep details. | Atomic model coordinates, structure factors (for X-ray), restraint files (for NMR). |
| Validation Reports | Includes map vs. model FSC, local resolution estimates, density fit. | Includes geometry (bond lengths/angles), clashscores, Ramachandran outliers. |
| Integrated Access | Accessed via EMDataResource (EMDR) with PDB. | PDB-Dev for integrative/hybrid models. Linked to EMDB for EM-derived models. |
| Resolution Range | Typically ~2 Å to >20 Å for virus particles. | Atomic resolution (≤ 1.0 Å) to medium resolution (~3-4 Å for cryo-EM). |
Table 2: Annual Deposition Trends (2020-2023) for Virus Structures
| Year | New EMDB Maps (Virus-related) | New PDB Depositions (EM method, Virus-related) | Notable Virus Example (Year Deposited) |
|---|---|---|---|
| 2020 | ~900 | ~450 | SARS-CoV-2 Spike Protein in situ (EMD-xxxxx, PDB-xxxx) |
| 2021 | ~950 | ~500 | Herpesvirus capsid portal complex (EMD-xxxxx) |
| 2022 | ~1000 | ~550 | HIV-1 Env trimer with broadly neutralizing antibodies |
| 2023 | ~1100 | ~600 | Norovirus VP1 particle bound to histo-blood group antigens |
Application Notes: The Deposition Workflow for a Virus Reconstruction
Note 1: Pre-deposition Curation. Before deposition, ensure your virus reconstruction meets community standards. For EMDB: final map file (CCP4/MRC format), accurate pixel size, resolution estimate (FSC 0.143 or 0.5 threshold), and detailed specimen preparation. For PDB: the fitted atomic model (PDBx/mmCIF format) must correspond to the EMDB map.
Note 2: Integrated Deposition via EMDataResource. The recommended pathway is the OneDep unified system. It guides the simultaneous deposition of the 3D map to EMDB and the associated atomic model to PDB, ensuring cross-referencing and consistency.
Note 3: Metadata Completeness. Comprehensive metadata is crucial for reuse. This includes: microscope and detector details, image processing software (e.g., RELION, cryoSPARC), symmetry imposed (e.g., icosahedral), and the genome sequence of the virus used.
Note 4: Post-Deposition Validation. Always review the automatically generated validation reports. For viruses, pay special attention to the map-model correlation in the capsid region and the geometry of protein-nucleic acid interactions.
Experimental Protocols
Protocol 1: Deposition of a Virus Cryo-EM Map to EMDB
Objective: To publicly archive a 3D reconstruction of an icosahedral virus capsid.
Materials:
- Final, sharpened cryo-EM map file (
.mrc). - Processing metadata (software versions, particle counts, etc.).
- Microscope and camera parameters.
- Sample and specimen preparation details.
Procedure:
- Prepare Map: Ensure your map is in MRC2014 format. Apply any recommended post-processing (masking, B-factor sharpening). Note the exact map half-sets if used for FSC.
- Access OneDep Portal: Navigate to the wwPDB OneDep system (https://deposit-pdbe.wwpdb.org/) and initiate a new EM deposition.
- Upload Map & Model: Upload the
.mrcfile. If an atomic model exists, upload the coordinate file. The system will link them. - Input Metadata: Complete all required fields in the multi-step form:
- Sample: Describe virus name, source, expressed system.
- Experiment: Specify imaging conditions (voltage, dose, detector, pixel size).
- Processing: Detail software, extracted particles, symmetry applied, final resolution.
- Map Information: Define origin, labeling of symmetry axes.
- Review & Submit: Preview the landing page and submit. An EMD-XXXXX accession code will be assigned after processing.
Protocol 2: Deposition of an Associated Atomic Model to PDB
Objective: To archive the atomic coordinates of a virus capsid protein fitted into a cryo-EM map.
Materials:
- Atomic model file (
.cifformat preferred). - Corresponding EMDB accession code.
- Sequence information of the modeled construct.
- Refinement and validation statistics.
Procedure:
- Prepare Model File: Convert model to PDBx/mmCIF format using
pdbx/mmciftools. Ensure chain IDs, residue numbering, and sequence are correct. - Coordinate Deposition: Within the same OneDep session as Protocol 1, or link to an existing EMDB deposition, provide the coordinate file.
- Define Polymer Sequences: Input or upload the precise amino acid/nucleotide sequence for each chain. Match the sequence to the construct used.
- Describe Refinement: Specify the refinement program (e.g., PHENIX, Refmac), the map used (EMD-ID), and restraints.
- Validation & Finalization: The system will run validation checks. Review the reports, address major outliers if possible, and finalize the submission to receive a PDB-XXXX code.
Title: Virus Structure Deposition Workflow via OneDep
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Virus Cryo-EM and Deposition
| Item | Function in Virus 3D Reconstruction | Example/Supplier |
|---|---|---|
| Quantifoil R2/2 Au Grids | Holey carbon grids with gold support, preferred for virus particles due to better stability and conductivity. | Quantifoil GmbH |
| Vitrobot Mark IV | Automated plunge-freezer for consistent vitrification of virus samples in a thin layer of amorphous ice. | Thermo Fisher Scientific |
| 300 kV Cryo-TEM | High-end transmission electron microscope (e.g., Krios, Glacios) providing high signal-to-noise images of frozen-hydrated viruses. | Thermo Fisher Scientific, JEOL |
| Direct Electron Detector | Camera (e.g., K3, Falcon 4) that counts individual electrons, enabling high-resolution, movie-based data collection. | Gatan, Thermo Fisher Scientific |
| RELION/cryoSPARC | Software suites for high-performance computing in single-particle analysis, including 3D classification and refinement. | MRC LMB, Structura Biotechnology Inc. |
| Coot | Interactive model-building software for fitting and adjusting atomic models into cryo-EM density maps. | MRC LMB |
| PHENIX | Software suite for the automated and manual refinement of atomic models against cryo-EM maps. | UCLA, Lawrence Berkeley Lab |
| OneDep System | The mandatory, unified online deposition system for public release of structures to EMDB and PDB. | wwPDB (EMDataResource) |
| EMRinger/PDB Validation | Key validation tools to assess model-to-map fit and model quality prior to deposition. | UCSF, wwPDB |
Conclusion
3D reconstruction of virus particles via cryo-EM has matured from a niche technique into a cornerstone of structural biology, offering unparalleled insights into viral architecture, life cycle, and host interactions. By understanding the foundational principles, meticulously executing the methodological pipeline, proactively troubleshooting common issues, and rigorously validating results against established metrics, researchers can generate reliable, high-resolution models. These models are not mere static pictures; they are dynamic blueprints that directly accelerate rational vaccine design, elucidate mechanisms of neutralization, and reveal novel druggable targets. Future directions point toward in situ tomography to visualize viruses within cells, time-resolved cryo-EM to capture dynamic processes, and the integration of AI/ML to automate processing and predict conformational states. For biomedical and clinical research, this continual advancement promises faster responses to emerging viral threats and a deeper mechanistic understanding of viral pathogenesis, fundamentally transforming our ability to develop targeted interventions.