From Cowpox to Code: A Technical History of Viral Vaccine Development for Biomedical Researchers
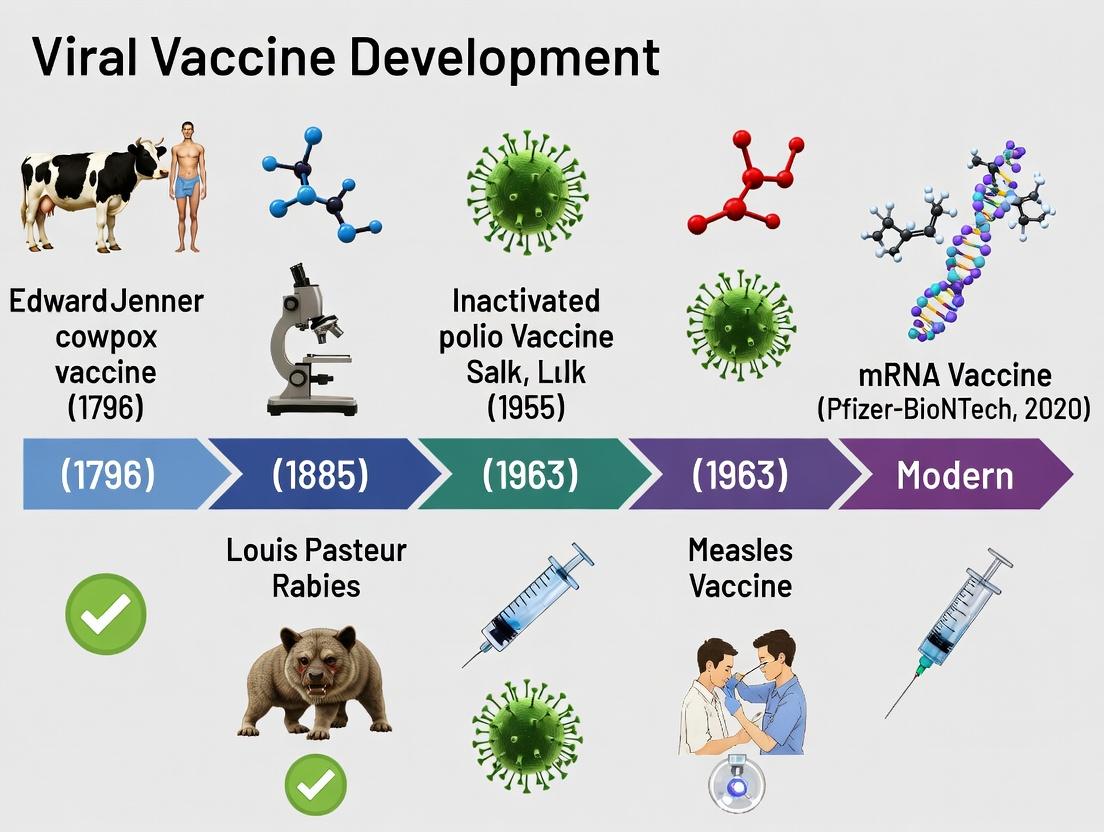
This article provides a comprehensive, technically detailed exploration of viral vaccine evolution, tracing the paradigm shifts from empirical observation to rational design.
From Cowpox to Code: A Technical History of Viral Vaccine Development for Biomedical Researchers
Abstract
This article provides a comprehensive, technically detailed exploration of viral vaccine evolution, tracing the paradigm shifts from empirical observation to rational design. Targeted at researchers, scientists, and drug development professionals, it examines the foundational principles established by pioneers like Jenner and Pasteur, details the methodologies of attenuated, inactivated, subunit, and vector platforms, and analyzes the troubleshooting and optimization challenges inherent to each. It concludes with a comparative validation of modern mRNA/LNP platforms against historical predecessors, synthesizing key learnings and outlining future directions for accelerated, precision vaccinology.
Empirical Beginnings to Cell Culture: The Foundational Paradigms of Early Virology
Application Notes
The 1796 inoculation experiment conducted by Edward Jenner represents the foundational empirical prototype for prophylactic vaccination. By demonstrating that deliberate infection with the mild cowpox virus (Vaccinia virus) conferred protection against the severe and often fatal human smallpox virus (Variola virus), Jenner established the principle of cross-protection. This principle—whereby immunity to one pathogen provides immunity to a related, more virulent pathogen—is a cornerstone of virology and immunology that informed subsequent vaccine development for over two centuries. Within the thesis on the history of viral vaccine development, Jenner's work is the pivotal origin point, predating the understanding of viruses or the immune system. Its legacy is the conceptual framework of using a biologically related but attenuated agent to safely induce protective immunity, a logic that extends from live-attenuated vaccines (e.g., Sabin polio, MMR) to modern vector-based and mRNA platforms which mimic this antigenic presentation without using the pathogenic organism itself.
Experimental Protocols
Protocol: Jenner's 1796 Cowpox Inoculation & Challenge Experiment
Objective: To test the hypothesis that prior infection with cowpox protects against subsequent infection with smallpox.
Materials:
- Subject: James Phipps, a healthy 8-year-old boy with no prior history of smallpox or cowpox.
- Inoculum Source: Fresh cowpox pustule material from the hand of milkmaid Sarah Nelmes.
- Inoculation Instrument: A lancet.
- Challenge Material: Fresh smallpox (Variola) pus taken from a patient with active disease.
Procedure:
- Pre-inoculation Observation: Confirm the subject is in good health with no signs of active disease.
- Cowpox Inoculation (Day 0): a. Make two superficial incisions, each about half an inch long, on the subject's left arm. b. Gently introduce the cowpox pustular matter into the incisions. c. Lightly dress the site.
- Post-Inoculation Monitoring (Days 1-9): a. Monitor the inoculation site daily for signs of reaction. b. Document the local progression: redness, vesicle formation, pustule development, and eventual scabbing. c. Monitor the subject for systemic symptoms (e.g., fever, axillary lymphadenopathy).
- Recovery Phase (Days 10-48): Allow the local lesion to fully resolve, confirming a typical, mild cowpox infection course.
- Smallpox Challenge (Day 49, July 1, 1796): a. Obtain fresh smallpox matter from a natural smallpox pustule. b. Inoculate the subject via multiple superficial incisions on both arms, using the same technique as in Step 2. c. Introduce the smallpox matter into the incisions.
- Challenge Phase Monitoring (Days 50-56+): a. Observe the challenge sites meticulously several times daily. b. Document the presence or absence of the characteristic "take" – the development of a Jennerian vesicle/pustule indicating successful smallpox infection. c. Monitor closely for any signs of systemic smallpox disease.
Interpretation: The absence of a disease "take" or systemic illness following the deliberate smallpox challenge was interpreted as successful protection conferred by the prior cowpox infection.
Modern Protocol:In VitroAssessment of Cross-Neutralizing Antibodies (Illustrative)
Objective: To demonstrate the cross-reactive immune response principle underlying Jenner's observation using modern virological techniques.
Materials: Cell culture (e.g., Vero cells), live Vaccinia virus (cowpox surrogate), live Variola virus (or a closely related orthopoxvirus like Monkeypox virus under appropriate biosafety conditions), serum from Vaccinia-immunized individuals, cell culture media, plaque assay reagents (crystal violet, formaldehyde).
Procedure:
- Serum Collection & Heat-Inactivation: Collect serum from a Vaccinia-immunized host. Heat-inactivate at 56°C for 30 minutes to degrade complement.
- Virus-Serum Mixture Preparation: a. Prepare serial dilutions of the inactivated serum (e.g., 1:10, 1:50, 1:100, 1:500) in sterile media. b. Mix a fixed, known titer (e.g., 1000 plaque-forming units, PFU) of Vaccinia virus with each serum dilution. Set up a parallel set of mixtures using Variola virus. c. Incubate mixtures at 37°C for 1 hour to allow antibody neutralization.
- Plaque Reduction Neutralization Test (PRNT): a. Infect confluent monolayers of Vero cells in multi-well plates with the virus-serum mixtures. b. Incubate to allow viral adsorption. c. Overlay with a semi-solid medium (e.g., carboxymethylcellulose) to restrict viral spread to neighboring cells. d. Incubate for 48-72 hours until plaques (clear zones of dead cells) form.
- Staining & Quantification: a. Fix cells with formaldehyde and stain with crystal violet. b. Count the number of plaques in each well.
- Analysis: Calculate the percentage reduction in plaque count for each serum dilution compared to virus-only controls. Determine the PRNT50 titer (serum dilution that reduces plaques by 50%).
Interpretation: The presence of a high PRNT50 titer against both Vaccinia and Variola viruses in the Vaccinia-immune serum provides in vitro validation of the cross-protective humoral immunity observed empirically by Jenner.
Data Presentation
Table 1: Key Outcomes from Jenner's 1796 Experiment and Modern Correlates
| Parameter | Jenner's 1796 Observation (Qualitative) | Modern Quantitative/Mechanistic Correlate |
|---|---|---|
| Primary Inoculation Agent | Cowpox pustule matter | Vaccinia virus (Orthopoxvirus) |
| Local Reaction to Inoculation | Vesicle at site, progressing to pustule & scab over ~9 days | Local viral replication, immune cell infiltration; lesion score 2-4 (scale 0-5) |
| Systemic Symptoms Post-Inoculation | Mild fever, discomfort | Documented in ~10-20% of primary vaccinees; fever >38°C |
| Time to Immunity | Challenge performed 7 weeks post-inoculation | Neutralizing antibodies detectable by 14 days post-primary vaccination, peak at 4-6 weeks |
| Challenge Agent | Smallpox (Variola) pustule matter | Variola virus (Orthopoxvirus) |
| Result of Challenge | No "take" or disease; minor local redness only | PRNT50 antibody titers >1:40 correlate with protection; cross-reactive T-cell responses measurable |
| Protective Efficacy | 100% in index case (n=1) | ~95% efficacy for Vaccinia against Variola in population studies |
Table 2: Research Reagent Solutions for Orthopoxvirus Cross-Protection Studies
| Reagent / Material | Function in Research | Example / Specification |
|---|---|---|
| Vaccinia Virus (e.g., strain Lister, NYCBH) | Live-attenuated vaccine strain; serves as the immunizing agent and a source of cross-reactive antigens. | GMP-produced, titered stock for in vivo immunization. |
| Variola Virus Surrogate (e.g., Monkeypox virus, Vaccinia virus expressing Variola antigens) | Used for challenge or neutralization assays under BSL-2/3 conditions to model smallpox immunity. | Recombinant viruses for specific antigenic focus. |
| Plaque Assay Reagents (Carboxymethylcellulose, Crystal Violet) | Enables quantification of infectious virus titers and neutralization antibody potency via PRNT. | Standard cell culture protocol reagents. |
| Orthopoxvirus-Specific ELISA Kits | Quantifies antigen-specific IgG/IgM antibody responses to Vaccinia and cross-reactive epitopes. | Commercial kits for vaccinia IgG, EEV/IMV antigens. |
| Flow Cytometry Antibody Panels (Mouse/Human) | Profiles cellular immune responses (CD4+, CD8+ T-cells, memory subsets) post-immunization. | Fluorochrome-labeled antibodies to CD3, CD4, CD8, CD44, CD62L, IFN-γ, TNF-α. |
| Mouse Models (e.g., BALB/c, C57BL/6) | In vivo models for studying vaccine efficacy, pathogenesis, and immune correlates of protection. | Defined pathogen-free, specific age/weight. |
Visualizations
Diagram 1: Jenner's empirical prototype and modern immunological principle of cross-protection.
Diagram 2: Workflow for plaque reduction neutralization test to measure cross-reactive antibodies.
Application Notes
AN-001: Historical Context and Conceptual Shift The development of a rabies vaccine by Louis Pasteur and Émile Roux in 1885 represents the first deliberate, laboratory-based attenuation of a pathogen to create a vaccine. Unlike Jenner's empirical use of cross-reactive cowpox, Pasteur's work was founded on a rational principle: serial passage and modification of an infectious agent could reduce its virulence while maintaining its immunogenicity. This established the paradigm of "isolate, inactivate/infectivate, and immunize" that defined virology and vaccine development for the next century, directly leading to methods for polio, measles, mumps, and rubella vaccines. It marks the transition from observational to experimental immunology.
AN-002: Quantitative Analysis of the 1885 Clinical Trial Pasteur's initial human trial, while not a controlled modern study, provided the first quantitative evidence of the vaccine's efficacy against a uniformly fatal disease.
Table 1: Outcomes from Pasteur's Initial Rabies Vaccine Administration (1885-1886)
| Patient Cohort | Number Treated | Deaths from Rabies | Reported Survival Rate | Historical Case Fatality Rate |
|---|---|---|---|---|
| Joseph Meister (first patient) | 1 | 0 | 100% | ~100% |
| First 350 patients treated post-exposure | 350 | 1 | 99.7% | ~100% |
| Russian patients (19 treated, 1886) | 19 | 3 | 84.2% | ~100% |
Key Protocol: The "Pasteur Protocol" for Post-Exposure Prophylaxis (PEP)
Principle: Progressive immunization starting with fully attenuated (desiccated) spinal cord material, followed by inoculations with material of increasing virulence, culminating in injections of fully virulent virus. This aimed to induce immunity before the virus, which has a long and variable incubation period, could reach the central nervous system.
Materials:
- Source of Virus: Rabies virus-infected rabbit spinal cords.
- Attenuation Agent: Desiccation over potassium hydroxide pellets in a sterile flask.
- Duration of Attenuation: Variable, from 14 days (fully attenuated) to 1-3 days (partially virulent).
- Vehicle: Sterile broth or saline for homogenization.
- Administration Route: Subcutaneous injection.
Detailed Methodology:
- Virus Propagation: Infect a rabbit intracerebrally with the "fixed" (laboratory-adapted) rabies virus (RV) strain. Monitor for paralytic symptoms.
- Harvesting: Upon the rabbit's death from rabies, aseptically remove the spinal cord.
- Attenuation: Suspend the cord in a sterile, dry flask above KOH pellets. Seal the flask.
- Preparation of Inoculum: For each vaccination, homogenize a segment of the desiccated cord in a sterile broth to create a suspension.
- Immunization Schedule (Standard 14-Day Protocol):
- Day 1-14: Administer daily subcutaneous injections.
- Day 1 & 2: Inject material desiccated for 14 days.
- Day 3 & 4: Inject material desiccated for 10-12 days.
- Day 5 & 6: Inject material desiccated for 7-8 days.
- Day 7-14: Inject progressively fresher (less desiccated) material, culminating in cord desiccated for only 1-3 days.
- Monitoring: Observe the patient for local reaction and for signs of rabies. The protocol was later modified (e.g., 10-day, 7-day) based on exposure severity.
The Scientist's Toolkit: Research Reagent Solutions for Historical Rabies Vaccine Research
Table 2: Key Research Materials and Their Functions
| Item | Function/Role in Experiment |
|---|---|
| Fixed Rabies Virus (RV) Strain | A virus serially passaged in rabbits to standardize incubation time (6-7 days) and pathogenicity, enabling reproducible experiments. |
| Rabbit Model (Oryctolagus cuniculus) | The primary in vivo system for virus propagation, virulence testing, and vaccine preparation. |
| Potassium Hydroxide (KOH) Pellets | A powerful desiccant. Controlled drying was the empirical method for viral attenuation, reducing infectivity while preserving antigenicity. |
| Sterile Broth (Nutrient Medium) | Vehicle for homogenizing spinal cord tissue to create injectable suspensions of the viral antigen. |
| Glass Flasks & Syringes | Essential for maintaining sterile conditions during tissue desiccation and injection, a critical advancement from Jenner's era. |
Diagram 1: Pasteur's Rabies Vaccine Dev Workflow
Diagram 2: Rational Attenuation Principle Logic
Diagram 3: Pathway from Jenner to mRNA Vaccines
Historical & Scientific Context
The cultivation of viruses in the embryonated chicken egg, pioneered by Ernest William Goodpasture and colleagues in the early 1930s, represents a pivotal inflection point in the history of vaccinology. This innovation bridged the era of Jennerian empiricism and early tissue explants with the modern age of industrial-scale viral vaccine production. It provided the first reliable, sterile, and scalable system for propagating obligate intracellular human and animal viruses, directly enabling the development and mass production of vaccines against influenza and yellow fever. Within the broader thesis of vaccine history, the "Egg Era" stands as the critical technological platform that made widespread viral immunization feasible from the 1940s through the 21st century, preceding cell culture and molecular techniques.
Foundational Protocol: Goodpasture's Method for Virus Propagation in Embryonated Eggs
Principle: The developing chicken embryo provides a rich environment of diverse, susceptible tissues and cell types for viral growth. The chorioallantoic membrane (CAM), amniotic cavity, and allantoic cavity serve as accessible sites for inoculation and harvest.
Protocol (Adapted from Goodpasture et al., 1931-1932):
2.1 Materials Preparation:
- Eggs: Specific Pathogen Free (SPF) white Leghorn chicken eggs, incubated at 37.5°C ± 0.5°C and 55-65% relative humidity for 9-11 days to achieve desired embryo development.
- Candling Device: To visualize embryo and vascular structures.
- Disinfection: 70% ethanol or iodine solution.
- Drill: Manual egg drill or needle for creating inoculation port.
- Inoculum: Sterile-filtered virus suspension.
- Syringe: 1 mL tuberculin syringe with a 25-27 gauge needle, ½ to 1 inch in length.
- Sealing Material: Sterile glue, melted paraffin wax, or cellophane tape.
- Incubator: Maintained at 35-37°C, humidity-controlled.
2.2 Stepwise Procedure (CAM Inoculation):
- Candle & Mark: Candle egg to identify viable embryo and air sac. Mark the boundary of the air sac and a point on the side over the CAM where major blood vessels are absent.
- Disinfect: Swab the marked area thoroughly with 70% ethanol.
- Create Inoculation Port: Using a sterile drill or needle, carefully pierce the shell at the marked site on the side (not over the air sac). Avoid puncturing the underlying shell membrane.
- Create Air Sac Vent (Optional): A small hole may be drilled over the air sac to create a negative pressure differential.
- Inoculate: Place the needle through the side port, gently penetrating the CAM. Deposit 0.1-0.5 mL of inoculum onto the CAM.
- Seal: Seal both holes (inoculation port and air sac vent) with sterile tape or wax.
- Incubate: Place eggs horizontally (with inoculation site upwards) in the incubator for the virus-specific duration (typically 2-5 days).
- Harvest: Refrigerate eggs at 4°C for 4-12 hours to euthanize embryo and constrict blood vessels. Re-disinfect, open shell over the air sac, and carefully dissect away the shell over the CAM. Excise the CAM using sterile forceps and scissors. Harvest viral material (pocks on CAM, or fluids from allantoic/amniotic cavities).
Application to Vaccine Development: Key Protocols & Data
Influenza Vaccine (Inactivated, Split-Virion) Production
Workflow Principle: Seed influenza virus strains (A or B) are adapted to grow in the allantoic cavity. Replication yields high-titer viral harvest, which is inactivated, purified, and disrupted.
Detailed Protocol for Virus Propagation:
- Egg Preparation: Use 9-11 day old embryonated SPF eggs.
- Inoculation (Allantoic Route): Candle and mark a point just above the air sac boundary. Disinfect. Drill a small hole. Direct a needle vertically (or at a slight angle) through the hole, piercing ~1.5-2 cm into the egg to reach the allantoic cavity. Inject 0.1-0.2 mL of seed virus (typically ~10^3-10^4 EID50/egg).
- Incubation: Incubate at 33-35°C (for human isolates) for 48-72 hours.
- Harvest: Chill eggs. Aseptically puncture the shell over the air sac, peel back, and puncture the underlying membranes. Use a sterile pipette or cannula to aspirate the allantoic fluid (typically 8-10 mL per egg). Pool fluids from multiple eggs.
- Clarification & Inactivation: Clarify pooled fluid by low-speed centrifugation. Inactivate virus using beta-propiolactone (BPL) or formaldehyde under controlled conditions (e.g., 1:4000 BPL at 4°C for 48-72 hours).
- Purification & Disruption: Purify via sucrose density gradient ultracentrifugation. Disrupt viral membrane using ether or detergent (e.g., Triton X-100) to produce "split-virion" antigen.
Quantitative Yield Data (Typical Modern Process):
Table 1: Influenza Vaccine Yield per Embryonated Egg
| Parameter | Typical Yield Range | Notes |
|---|---|---|
| Allantoic Fluid Volume | 8 - 12 mL | Depends on egg age and incubation conditions. |
| Viral Titer (HA Units) | 128 - 1024 HAU/50 μL | Hemagglutination assay titer of harvest pool. |
| Egg Infectious Dose (EID50) | 10^8 - 10^9 /mL | Titer of harvested allantoic fluid. |
| Antigen Yield per Egg | ~1 - 5 μg HA | Yield of purified hemagglutinin (HA) antigen after processing. |
| Doses per Egg | ~0.5 - 1.5 | Approximate number of standard monovalent doses (15 μg HA/dose). |
Diagram 1: Influenza Vaccine Production in Eggs
17D Yellow Fever Vaccine Production (Live-Attenuated)
Workflow Principle: The attenuated 17D-204 strain is propagated in the embryonated egg, specifically in embryo tissue, to produce a live viral vaccine.
Detailed Protocol for 17D Vaccine Production:
- Egg Preparation: Incubate SPF eggs for 7-8 days.
- Inoculation (Embryonic Tissue): Candle and mark a point on the shell directly over the embryo. Disinfect. Drill a small window (~1 cm diameter). Carefully remove the shell piece without damaging the shell membrane. Apply a drop of sterile saline to the membrane, which is then torn open with sterile forceps. Inoculate 0.1-0.2 mL of 17D seed virus directly onto the embryo.
- Sealing & Incubation: Seal the window with sterile tape or a plastic film. Incubate horizontally at 37°C for 3-4 days.
- Harvest: Chill eggs. Aseptically open the window and harvest the entire embryo (excluding eyes and beak) and any surrounding fluid into a sterile container. Homogenize the embryo tissue in a suitable buffer (e.g., sterile water or stabilizer) to create a suspension.
- Clarification & Stabilization: Clarify the homogenate by centrifugation. The supernatant containing the virus is stabilized with excipients (e.g., sorbitol, gelatin) and lyophilized for long-term storage.
Quantitative & Quality Control Data:
Table 2: 17D Yellow Fever Vaccine Production & QC Specifications
| Parameter | Typical Target/Value | Purpose/Notes |
|---|---|---|
| Inoculum Titer | ≥ 3.0 log10 PFU/0.2mL | Seed virus potency. |
| Incubation Period | 72 - 96 hours | Optimized for viral yield. |
| Viral Yield per Embryo | 3.0 - 5.0 log10 PFU/mL | In final homogenate. |
| Final Dose Potency | ≥ 3.0 log10 IU per dose | WHO minimum requirement (Post-lyophilization). |
| Neurovirulence Test (MNT) | Must pass | In vivo test in mice to ensure attenuation. |
| Immunogenicity (Seroconversion) | ≥ 90% in recipients | Primary efficacy endpoint. |
Diagram 2: 17D Yellow Fever Vaccine Production
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents & Materials for Egg-Based Virology/Vaccinology
| Item | Function & Application | Key Notes |
|---|---|---|
| SPF Embryonated Eggs | Virus propagation substrate. Must be free of specific avian pathogens to avoid contamination and interference. | Foundation of the system; defines sterility and yield. |
| Beta-Propiolactone (BPL) | Alkylating agent for viral inactivation. Denatures nucleic acids while preserving antigenic structure of coat proteins. | Preferred for influenza vaccines over formaldehyde due to less antigen modification. |
| Sucrose (Density Gradient Grade) | Medium for rate-zonal or equilibrium ultracentrifugation. Separates whole virions from cellular debris and subviral particles. | Critical for purifying virus from allantoic fluid. |
| Triton X-100 (or similar detergent) | Non-ionic surfactant used to disrupt the viral lipid envelope, creating a "split-virion" vaccine. | Reduces reactogenicity compared to whole-virion inactivated vaccines. |
| Sorbitol-Gelatin Stabilizer | Stabilizing excipient for live-attenuated vaccines (e.g., YF 17D). Protects viral potency during lyophilization and storage. | Prevents loss of infectivity/titer due to heat or freeze-drying stress. |
| Specific Antisera (e.g., Anti-HA) | Used in immunochemical assays (HI, SRH) to quantify and characterize vaccine antigens. | Essential for lot potency testing and antigenic characterization. |
| Trypsin-TPCK | Serine protease inhibitor-treated trypsin. Used to cleave and activate influenza HA protein in cell-based assays (e.g., MDCK cells) but sometimes used in egg adaptation studies. | Facilitates multi-cycle replication in vitro for certain virus strains. |
Contemporary Relevance & Limitations
The embryonated egg remains the dominant global platform for seasonal influenza vaccine manufacturing due to its high yield, proven safety record, and established regulatory pathways. However, limitations include: the potential for egg-adaptive mutations in the influenza HA gene that can alter antigenicity ("egg-passage effects"); production timelines of several months; and scalability challenges during pandemics. These limitations have driven the parallel development of cell-culture and recombinant (e.g., baculovirus/insect cell) platforms. Nevertheless, the egg-based system is a cornerstone technology that successfully transitioned vaccinology from a cottage industry to a global public health enterprise, directly saving millions of lives and establishing the biological principles for subsequent viral vaccine development.
The 1949 publication by John F. Enders, Thomas H. Weller, and Frederick C. Robbins, detailing the successful cultivation of poliovirus in non-neural human tissue, represents a pivotal turning point in virology and vaccine development. Situated within the broader history from Jenner’s empirical inoculations to contemporary mRNA platforms, this work dismantled the prevailing “neurotropic” dogma of poliovirus. By demonstrating that the virus could be grown in vitro in cultures of human embryonic skin, muscle, and intestinal tissue, they provided the essential, reproducible, and scalable platform necessary for modern virology. This breakthrough directly enabled the isolation, characterization, and attenuation of viruses, paving the way for the development of the Salk (inactivated) and Sabin (live-attenuated) polio vaccines and establishing the standard methodology for subsequent viral vaccine research against measles, mumps, rubella, and varicella.
Key Quantitative Findings from the 1949 Study
Table 1: Summary of Successful Poliovirus Cultivation in Various Tissues
| Tissue Type (Human Embryonic) | Virus Strain(s) Tested | Evidence of Viral Growth (Cytopathic Effect) | Key Outcome for Vaccine Development |
|---|---|---|---|
| Skin & Muscle (Miscellaneous) | Lansing, Leon, Brunhilde | Yes (Tissue degeneration) | Proof of principle: virus multiplies in non-neural cells. |
| Intestinal Tissue | Brunhilde | Yes | Suggested enteric replication site, crucial for understanding transmission. |
| Suspended Tissue Fragments in Nutrient Medium | Lansing | Yes - maintained over several passages | Established serial propagation, enabling large-scale virus production. |
| Control Tissues (Uninfected) | N/A | No degeneration | Confirmed that degeneration was virus-specific. |
Table 2: Impact Metrics of the In Vitro Breakthrough
| Metric | Pre-1949 (Neural Tissue Only) | Post-1949 (Non-Neural Cell Culture) | Consequence |
|---|---|---|---|
| Virus Production Scale | Limited, cumbersome (monkey brain/spinal cord) | Virtually unlimited, reproducible | Enabled industrial-scale vaccine manufacturing. |
| Research Safety | High risk (working with neural tissue, live animals) | Significantly reduced risk (contained cell cultures) | Made virology labs safer and more accessible. |
| Speed of Isolation/Identification | Slow (weeks to months, relying on animal symptoms) | Rapid (days, observing cell monolayers) | Accelerated viral discovery and diagnostic capability. |
| Genetic Stability of Virus | Difficult to assess | Easy to monitor and control through serial passage | Enabled rational attenuation for live vaccines (e.g., Sabin). |
Detailed Experimental Protocols
Protocol 1: Preparation of Roller Tube Cultures from Human Embryonic Tissue
This is a detailed reconstruction of the core methodology from the 1949 paper.
I. Reagent & Material Preparation
- Balanced Salt Solution (BSS): Prepare a sterile solution of inorganic salts (NaCl, KCl, CaCl₂, NaHCO₃, NaH₂PO₄) in distilled water. This serves as a washing and base medium.
- Plasma-Clot Substrate: Prepare a mixture of chicken plasma and embryonic extract (typically from chick embryos) in Hanks' BSS. This forms the coagulated "clot" that supports tissue fragment attachment.
- Nutrient Maintenance Medium: A mixture of:
- Human serum (inactivated at 56°C for 30 minutes)
- Balanced Salt Solution
- Embryonic extract
- Penicillin & Streptomycin (a critical advancement used by Enders et al. to prevent bacterial contamination).
II. Tissue Collection and Fragmentation
- Aseptically obtain human embryonic tissue (e.g., skin, muscle, intestine) of approximately 4-5 months gestation.
- Immediately place tissue in cold BSS.
- Using sterile instruments and a Petri dish, meticulously mince the tissue into fragments approximately 1-2 mm³ in size.
- Wash the fragments thoroughly with several changes of cold BSS to remove blood cells and debris.
III. Explant Cultivation in Roller Tubes
- Place a small drop of chicken plasma onto the inner wall of a sterile Pyrex test tube (e.g., 16 x 150 mm).
- Add 2-3 tissue fragments to the plasma drop.
- Add one drop of embryonic extract and gently tilt the tube to mix, allowing the mixture to coagulate and entrap the fragments.
- After the clot forms (5-10 minutes), add 1.0 - 1.5 mL of the nutrient maintenance medium to the tube.
- Cap the tube loosely and place it in a roller drum apparatus at 36-37°C, rotating at approximately 8-12 revolutions per hour.
IV. Virus Inoculation and Observation
- After 24-48 hours of incubation (allowing tissue outgrowth), decant the original nutrient medium.
- Inoculate the culture with 0.1-0.2 mL of a clarified suspension of virus (e.g., Lansing strain from infected mouse brain).
- Allow the virus to adsorb for 30-60 minutes at room temperature.
- Add fresh maintenance medium and return the tube to the roller drum.
- Daily Examination: Observe cultures microscopically (50-100x magnification) for signs of specific cytopathic effect (CPE), primarily the degeneration and disintegration of fibroblastic cells growing from the tissue fragments.
- Maintain cultures by changing half the medium every 3-4 days.
V. Virus Passage
- Upon observation of advanced CPE (usually 5-7 days post-inoculation), harvest the culture by freezing and thawing the entire tube contents.
- Clarify the supernatant by light centrifugation.
- Use this supernatant to inoculate fresh, healthy tissue cultures to demonstrate serial transmission of the infectious agent.
Protocol 2: Titration of Viral Infectivity in Tissue Culture (TCID₅₀)
This endpoint assay, enabled by their work, became the gold standard for quantifying virus.
I. Preparation of Serial Virus Dilutions
- Prepare tenfold serial dilutions (10⁻¹ to 10⁻⁸) of your virus stock in maintenance medium or a suitable diluent (e.g., BSS with 2% serum).
- Use fresh pipettes or tips for each dilution to ensure accuracy.
II. Inoculation of Culture Plates or Tubes
- Prepare replicate cell culture wells or tubes (e.g., 8-10 per dilution) containing confluent or near-confluent monolayers of susceptible cells (e.g., primary monkey kidney, HeLa, or fibroblast cells).
- Aspirate the growth medium from all wells/tubes.
- Inoculate each well/tube in a dilution series with an equal volume (e.g., 100 µL) of the corresponding virus dilution.
- Include control wells/tubes inoculated with diluent only.
III. Incubation and Observation
- Allow adsorption for 1-2 hours at 37°C, then add maintenance medium.
- Incubate cultures at 37°C and observe daily for CPE under a microscope.
- Record for each well/tube whether it is positive (shows CPE) or negative (no CPE) at a time when the endpoint is clear (typically 5-7 days).
IV. Calculation of TCID₅₀ The 50% tissue culture infectious dose (TCID₅₀) is calculated using the method of Reed and Muench.
- Tally the cumulative number of positive and negative cultures at each dilution.
- Calculate the ratio of cumulative positives to total cultures at each dilution.
- Identify the two dilutions where the ratio brackets 50%.
- Use the formula: Log TCID₅₀ = X + (0.5 - P)/(P-N), where:
- X = Log of the dilution where the positive ratio is >50%.
- P = Proportion positive at dilution X.
- N = Proportion positive at the next higher dilution (where ratio is <50%).
Research Reagent Solutions & Essential Materials
Table 3: The Scientist's Toolkit for Mid-20th Century Cell Culture Virology
| Reagent/Material | Function in the Experiment | Modern Analog/Evolution |
|---|---|---|
| Human Embryonic Tissue Fragments | The primary explant providing living, susceptible cells for viral replication. | Continuous cell lines (e.g., Vero, MRC-5, HEK-293), primary cells, or induced pluripotent stem cell (iPSC)-derived tissues. |
| Chicken Plasma & Embryonic Extract | Provided the complex, undefined protein matrix ("clot") for cell attachment and initial growth factors. | Defined extracellular matrices (e.g., Matrigel, collagen, laminin) and serum-free, chemically defined media supplements. |
| Hanks' Balanced Salt Solution (BSS) | Maintained physiological pH and osmolarity for tissue washing and as a medium base. | Advanced buffers like Dulbecco's PBS (DPBS) and HEPES-buffered media for precise pH control. |
| Inactivated Human Serum | Supplied essential nutrients, hormones, and growth factors in the maintenance medium. | Fetal Bovine Serum (FBS) or, preferably, defined serum replacements for consistency and reduced variability. |
| Penicillin-Streptomycin | Critical innovation: Prevented bacterial and fungal contamination in long-term cultures. | Standard antibiotic/antimycotic cocktails (e.g., Pen-Strep-Amphotericin B) or use of strict aseptic technique in clean rooms. |
| Roller Drum Apparatus | Continuously bathed tissue fragments in medium and improved gas exchange, promoting better cell health than static cultures. | CO₂ incubators for static plate/tube culture, or sophisticated bioreactors and wave bags for large-scale suspension culture. |
| Pyrex Test Tubes | The primary culture vessel. | Multi-well plastic plates (6-384 wells), T-flasks, roller bottles, and cell factories for high-throughput applications. |
Visualizations
Title: In Vitro Poliovirus Cultivation Workflow (1949)
Title: Impact of Cell Culture Breakthrough on Virology
Application Notes
The development of poliovirus vaccines represents a foundational case study in the history of virology, illustrating the strategic application of cell culture systems to create two distinct vaccine paradigms: the inactivated (Salk) and live-attenuated (Sabin) vaccines. These approaches, born in the mid-20th century, directly informed subsequent vaccine development for other pathogens, including measles. The use of primary monkey kidney cells for poliovirus propagation was a critical technological leap, moving vaccine production away from animal models and into controlled in vitro systems. This shift enabled both the large-scale virus production needed for inactivation and the sequential passage required for empirical attenuation.
The success of these paradigms hinged on understanding viral tropism and replication kinetics in cultured cells. The Salk vaccine’s safety depended on complete chemical inactivation without compromising immunogenicity, a delicate balance. The Sabin vaccine relied on the selection of stable, attenuated mutants that retained the capacity for limited replication in the human gut to induce robust mucosal immunity, but not neurovirulence. The measles vaccine (Edmonston strain), developed by Enders and colleagues, followed the Sabin paradigm, using serial passage in chick embryo cells to achieve attenuation. These historical milestones, situated between Jenner’s empiricism and today’s rational design, established cell culture as the indispensable workhorse of classic vaccinology.
Protocols
Protocol 1: Primary Monkey Kidney Cell Culture for Poliovirus Propagation (Historical Method)
Objective: To prepare primary cell cultures for the initial isolation and large-scale production of poliovirus for both Salk and Sabin vaccine development.
Materials:
- Rhesus or cynomolgus monkey kidneys.
- Hanks' Balanced Salt Solution (HBSS) with antibiotics (Penicillin 100 U/mL, Streptomycin 100 µg/mL).
- 0.25% Trypsin solution.
- Growth Medium: Mixture 199 or Eagle's Basal Medium (BME) supplemented with 2-5% calf serum.
- Maintenance Medium: As above, but with reduced serum (0.5-1%).
- Sterile dissection tools, magnetic stirrer with warming plate, centrifuge.
- Tissue culture flasks or roller bottles.
Procedure:
- Aseptically remove kidneys from sacrificed monkeys. Decapsulate and mince cortical tissue into ~1-2 mm³ fragments.
- Wash tissue fragments 3-4 times with cold HBSS to remove blood cells.
- Transfer tissue to a trypsinization flask. Add pre-warmed (37°C) 0.25% trypsin. Stir gently at 37°C for 15-20 minutes.
- Discard the first supernatant containing debris. Continue sequential trypsinization cycles (10-15 minutes each), collecting the cell-rich supernatants in cold growth medium containing serum to inhibit trypsin.
- Pool the collected cell suspensions, filter through sterile gauze, and centrifuge at 150 x g for 10 minutes.
- Resuspend the cell pellet in growth medium. Count cells using a hemocytometer; viability should exceed 90% (Trypan Blue exclusion).
- Seed culture vessels at a density of 2-3 x 10⁵ cells/cm². Incubate at 37°C in a 5% CO₂ atmosphere.
- Once a confluent monolayer forms (typically 5-7 days), replace growth medium with maintenance medium. The monolayer is now ready for viral inoculation.
Virus Inoculation:
- Inoculate maintenance medium with poliovirus seed stock at a low multiplicity of infection (MOI ~0.1).
- Incubate until extensive cytopathic effect (CPE—cell rounding and detachment) is observed (24-72 hours).
- Harvest by freeze-thawing the culture vessel to release cell-associated virus. Clarify by low-speed centrifugation. The supernatant is the viral harvest.
Protocol 2: Serial Passage for Attenuation (Sabin Poliovirus & Edmonston Measles Virus Paradigm)
Objective: To empirically attenuate a wild-type virus by serial passage in non-human or unnatural host cell systems to select for variants with reduced human pathogenicity.
Materials:
- Wild-type virus stock (e.g., Poliovirus Mahoney type 1, Measles Edmonston wild-type).
- Non-human cell substrates (e.g., Primary Monkey Kidney for poliovirus; Chick Embryo Fibroblasts (CEF) for measles).
- Appropriate cell culture maintenance media.
- -80°C freezer or dry ice/ethanol bath for snap-freezing.
Procedure:
- Prepare confluent monolayers of the chosen foreign cell substrate in multiple tissue culture flasks.
- Passage 1: Inoculate the first flask with the wild-type human virus at a low MOI (~0.01). Incubate until moderate CPE is evident.
- Harvest the virus by freeze-thaw and clarification. This is the P1 (Passage 1) stock.
- Serial Passaging: Use a small, standardized volume (e.g., 0.1 mL) of the harvested virus lysate to inoculate the next fresh cell monolayer. Repeat this process for numerous passages (often 20-50+).
- Monitoring: Periodically, assess the biological phenotype of the passaged virus:
- Plaque Morphology: Compare plaque size and clarity to wild-type under agar overlay.
- Temperature Sensitivity: Replicate efficiency at lower (e.g., 32°C) vs. human body temperature (37°C).
- Animal Neurovirulence Testing (for poliovirus): Intracerebral inoculation of monkeys or transgenic mice to assess loss of neurovirulence (critical for Sabin strains).
- Clone the final attenuated population by plaque purification three times to ensure genetic homogeneity.
- Prepare a Master Seed Virus from a validated clone for vaccine production.
Protocol 3: Formalin Inactivation of Poliovirus (Salk Vaccine Paradigm)
Objective: To completely inactivate infectivity of poliovirus while preserving its antigenic integrity for use as a killed vaccine.
Materials:
- Clarified, high-titer poliovirus harvest from cell culture.
- 37% Formaldehyde solution.
- Phosphate-Buffered Saline (PBS), pH 7.2-7.4.
- Magnetic stirrer with temperature control (preferably cold room).
- Safety cabinet for handling infectious virus.
Procedure:
- Filter the virus harvest through a 0.45 µm filter to remove large cell debris.
- Transfer the virus suspension to an inactivation vessel. Place on a magnetic stirrer in a cold room (4°C) or controlled temperature water bath.
- Inactivation: Add formaldehyde to a final concentration of 1:4000 (0.025% v/v). Maintain constant, gentle agitation.
- Sampling and Safety Testing: Throughout the process, aseptically remove samples at defined intervals (e.g., day 1, 3, 5, 7...).
- Infectivity Test: Inoculate samples onto sensitive cell monolayers (e.g., HeLa or primary monkey kidney). Observe for CPE for at least 14 days. Test large volumes (e.g., 5 mL of inactivated sample per culture vessel) to detect low levels of residual live virus.
- Completion: Inactivation is considered complete and safe only when no live virus is detected in multiple consecutive samples taken after the theoretical inactivation endpoint. The original Salk protocol required a minimum of 9-12 days of treatment.
- Residual Formaldehyde Removal: After confirmation of complete inactivation, the formaldehyde may be neutralized with sodium bisulfite or removed via dialysis or tangential flow filtration.
- The inactivated viral antigen is then blended, sterile-filtered, and adjuvanted (commonly with alum for other inactivated vaccines, though not used in the original Salk vaccine).
Table 1: Key Quantitative Parameters of Poliovirus Vaccine Development
| Parameter | Salk (Inactivated) Vaccine | Sabin (Live-Attenuated) Vaccine | Notes |
|---|---|---|---|
| Cell Substrate | Primary Rhesus Monkey Kidney | Primary Monkey Kidney, then Human Diploid Cells (WI-38, MRC-5) | Shift to diploid cells addressed adventitious agent risk. |
| Virus Titer at Harvest (PFU/mL) | ~10⁸ – 10⁹ | ~10⁷ – 10⁸ (per strain) | High yield critical for Salk manufacturing scale. |
| Inactivation Agent & Concentration | Formaldehyde, 1:4000 (0.025%) | Not Applicable | Critical parameter for safety/immunogenicity balance. |
| Inactivation Duration | 9-12 days at 37°C | Not Applicable | Safety tested by extended cell culture observation. |
| Passages to Attenuation | Not Applicable | 52-55 passages (Sabin type 1 in monkey cells) | Empirical process selecting for neuro-attenuating mutations. |
| Doses per Regimen | 3-4 intramuscular injections | 3 oral doses | Sabin induces stronger mucosal immunity in gut. |
| Vaccine Efficacy | 80-90% against paralytic disease | >95% against infection and disease | Sabin more effective at interrupting transmission. |
Table 2: Research Reagent Solutions for Cell Culture-Based Vaccine Development
| Reagent / Material | Function in Protocol | Critical Notes |
|---|---|---|
| Primary Monkey Kidney Cells | Permissive substrate for poliovirus isolation and initial propagation. | Source of adventitious agents (e.g., SV40); replaced by diploid cells. |
| Chick Embryo Fibroblasts (CEF) | Foreign cell substrate for attenuating measles virus (Edmonston strain). | Non-human cells select for virus variants less adapted to human hosts. |
| Human Diploid Cell Lines (WI-38, MRC-5) | Standardized, characterized substrate for production of attenuated viruses (Sabin, measles, rubella, etc.). | Finite lifespan, free of adventitious agents, ensures safety and consistency. |
| Trypsin Solution (0.25%) | Enzymatic dissociation of tissue to establish primary cell cultures. | Quality and sterility are paramount to prevent culture contamination. |
| Serum (Calf/Bovine/Fetal Bovine) | Provides essential growth factors and nutrients for cell proliferation. | Batch variability and risk of bovine contaminants require careful screening. |
| Maintenance Medium (Low Serum) | Supports cell viability while permitting efficient viral replication. | Low serum reduces interference with virus adsorption and harvest. |
| Formaldehyde (37% Solution) | Alkylating agent for chemically inactivating viral infectivity. | Concentration, temperature, and duration are critical validated parameters. |
| Agarose / Methylcellulose Overlay | For plaque assays to titer virus or isolate pure clones. | Immobilizes virus to form distinct plaques for counting and picking. |
Visualizations
Platform Evolution: Technical Methodologies from Subunit Vaccines to Viral Vectors
The development of the Hepatitis B Virus (HBV) vaccine using recombinant DNA technology in the 1980s represents a pivotal inflection point in the history of immunization, bridging the era of empiric whole-pathogen vaccines (Jenner's smallpox) to the rational design of modern subunit and nucleic acid platforms (mRNA). For the first time, a viral immunogen was produced not by cultivating the infectious agent, but by instructing a microbial factory (Saccharomyces cerevisiae) to manufacture a specific, safe, and protective antigen: the Hepatitis B Surface Antigen (HBsAg). This Application Note details the key protocols and methodologies that enabled this revolution, providing a template for subsequent recombinant protein vaccines.
The traditional plasma-derived HBV vaccine, while effective, faced limitations in scalability, safety concerns regarding blood-borne pathogens, and public perception. The recombinant approach solved these challenges decisively.
Table 1: Comparison of Plasma-Derived vs. Recombinant HBsAg Vaccine
| Parameter | Plasma-Derived Vaccine (Heptavax-B) | Recombinant Vaccine (Recombivax HB) |
|---|---|---|
| Source of Antigen | Purified from plasma of chronically infected donors | Expressed in recombinant S. cerevisiae |
| Production Scale | Limited by donor plasma supply | Virtually unlimited microbial fermentation |
| Theoretical Safety Risk | Potential for live HBV or unknown blood-borne pathogens | No human-derived pathogens |
| HBsAg Yield | ~1-5 mg/L of source plasma | 1-10 mg/L of yeast culture (significantly scalable) |
| Key Adjuvant | Aluminum hydroxide | Aluminum hydroxide |
| Vaccine Efficacy | 90-95% in healthy adults | 90-95% in healthy adults |
| FDA Approval Year | 1981 | 1986 |
Table 2: Key Milestones in Recombinant HBsAg Vaccine Development
| Year | Milestone | Key Finding/Outcome |
|---|---|---|
| 1979 | HBsAg gene cloned | Viral DNA fragment encoding HBsAg inserted into E. coli plasmid. |
| 1981 | Expression in yeast demonstrated | HBsAg expressed in S. cerevisiae; forms virus-like particles (VLPs). |
| 1984 | Large-scale clinical trials begin | Demonstrated safety and immunogenicity equivalent to plasma vaccine. |
| 1986 | FDA licenses Recombivax HB (Merck) | First recombinant protein vaccine approved for human use. |
| 1987 | FDA licenses Engerix-B (GSK) | Second recombinant vaccine, using different S. cerevisiae strain/process. |
Core Protocols & Methodologies
Protocol 1: Cloning of the HBsAg Gene into an Yeast Expression Vector
Objective: To insert the gene encoding the major HBsAg (S protein, 226 amino acids) into a shuttle vector for transformation and expression in S. cerevisiae.
Materials:
- Source: HBV DNA genome (subtype ayw or adw).
- Restriction Enzymes: EcoRI, BamHI.
- Vector: pAO815-type yeast expression vector containing:
- Strong, inducible promoter (e.g., Alcohol Oxidase 1 - AOX1).
- Selectable marker (e.g., HIS4 gene for histidine prototrophy).
- Terminator sequence.
- Bacterial origin for propagation in E. coli.
- Host Cells: E. coli DH5α for cloning, S. cerevisiae strain (e.g., GS115 for Pichia pastoris system).
- Culture Media: LB + ampicillin; Minimal Dextrose (MD) plates lacking histidine for yeast selection.
Procedure:
- Gene Isolation: Digest HBV genomic DNA with appropriate restriction enzymes to excise the ~680 bp fragment encoding the HBsAg S region.
- Vector Preparation: Linearize the yeast expression vector with compatible restriction enzymes.
- Ligation: Ligate the HBsAg fragment into the linearized vector using T4 DNA ligase.
- Bacterial Transformation: Transform ligation mix into competent E. coli cells. Select transformants on LB-ampicillin plates.
- Plasmid Verification: Isolate plasmid DNA from bacterial colonies. Verify insert presence and orientation by restriction digest and DNA sequencing.
- Yeast Transformation: Linearize the verified plasmid within the HIS4 marker sequence. Transform into histidine-deficient (his4) S. cerevisiae via electroporation or chemical method.
- Selection: Plate transformed yeast on MD plates lacking histidine. Only cells with successful genomic integration of the vector (containing HIS4 and HBsAg gene) will grow.
Title: Cloning and Yeast Transformation Workflow for HBsAg
Protocol 2: Fermentation and Induction of HBsAg Expression inPichia pastoris
Objective: To produce HBsAg in a controlled bioreactor, inducing high-level expression under the AOX1 promoter.
Materials:
- Bioreactor: Fermenter with controls for pH, dissolved oxygen (DO), temperature, and agitation.
- Basal Salts Medium: Contains glycerol, salts, PTM1 trace elements.
- Induction Feed: Methanol (100% with PTM1 trace elements) as carbon source and AOX1 inducer.
- Antifoam Agent.
- Centrifuges and Cell Disruption Equipment.
Procedure:
- Fermentation Setup: Inoculate recombinant yeast clone into a basal salts/glycerol medium in a bioreactor.
- Glycerol Batch Phase: Grow cells to high cell density until glycerol is depleted (monitored by DO spike).
- Glycerol Fed-Batch Phase: Feed glycerol at a controlled rate to further increase biomass.
- Methanol Induction Phase: Initiate continuous feed of methanol once glycerol is fully consumed. The methanol serves as both carbon source and inducer for the AOX1 promoter, driving high-level HBsAg expression. Continue for ~72-100 hours.
- Harvest: Terminate fermentation. Centrifuge culture broth to separate cells from supernatant. For intracellular HBsAg (as in early processes), retain cell pellet.
- Cell Disruption: Lyse yeast cells using high-pressure homogenization or bead milling.
Title: Pichia pastoris Fermentation Phases for HBsAg Production
Protocol 3: Purification and VLP Assembly of Recombinant HBsAg
Objective: To purify HBsAg from yeast lysate and allow it to self-assemble into 22-nm Virus-Like Particles (VLPs), the immunogenic form.
Materials:
- Chromatography Systems: FPLC or equivalent.
- Chromatography Resins: Hydrophobic Interaction Chromatography (HIC) media (e.g., Phenyl Sepharose), Size Exclusion Chromatography (SEC) media (e.g., Sephacryl S-400).
- Buffers: Lysis buffer, high-salt binding buffer for HIC, low-salt elution buffer, formulation buffer.
- Ultrafiltration/Diafiltration (UF/DF) system.
- Analytical Tools: SDS-PAGE, Western Blot (anti-HBsAg), Electron Microscopy.
Procedure:
- Clarification: Clarify cell lysate by centrifugation and filtration to remove debris.
- Hydrophobic Interaction Chromatography (HIC): Adjust clarified lysate to high salt concentration. Load onto HIC column. HBsAg, being highly hydrophobic, binds strongly. Elute with a descending salt gradient. This step provides significant purification and concentration.
- VLP Assembly & Dialysis: Pool HIC eluates containing HBsAg. Dialyze or UF/DF into a low-salt, neutral pH formulation buffer. Under these conditions, HBsAg monomers spontaneously assemble into 22-nm VLPs.
- Size Exclusion Chromatography (SEC): Load assembled material onto SEC column. This step separates intact VLPs from aggregates and remaining impurities, yielding a monodisperse product.
- Sterile Filtration & Adsorption: Sterile filter the purified VLP preparation. Adsorb to aluminum hydroxide adjuvant (Alum) by mixing under controlled conditions.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Recombinant HBsAg Vaccine Development
| Reagent/Material | Function in HBsAg Vaccine Development |
|---|---|
| S. cerevisiae (Pichia pastoris) GS115 (his4-) | A methylotrophic yeast host strain; allows high-density fermentation, strong AOX1 promoter use, and selection via histidine prototrophy. |
| pAO815-based Expression Vector | Shuttle vector with AOX1 promoter and terminator, HIS4 marker; enables methanol-inducible, high-level expression in yeast. |
| Methanol (Induction Grade) | Serves as both carbon source and potent inducer of the AOX1 promoter, driving recombinant protein expression. |
| Phenyl Sepharose HIC Resin | Key purification resin exploiting the hydrophobic nature of HBsAg for capture and initial purification from crude lysate. |
| Sephacryl S-400 HR | Size exclusion chromatography medium for polishing step; isolates correctly assembled 22-nm VLPs from aggregates. |
| Aluminum Hydroxide Gel (Alum) | Adjuvant; adsorbs purified HBsAg VLPs, enhancing immune response by forming a depot and stimulating innate immunity. |
| Monoclonal Anti-HBsAg Antibodies | Critical for identity testing (Western Blot), quantification (ELISA), and monitoring purification yield and antigen integrity. |
The successful deployment of the recombinant HBsAg vaccine validated the entire paradigm of heterologous protein expression for human prophylaxis. It provided a scalable, safe, and efficacious product, directly addressing the limitations of its plasma-derived predecessor. This breakthrough laid the essential technical and regulatory groundwork for subsequent recombinant vaccines (HPV, shingles) and demonstrated the power of Virus-Like Particles—a concept now central to vaccinology. As a cornerstone in the thesis of vaccine evolution, it represents the critical transition from cultivating pathogens to programming genetic instructions, a principle that finds its ultimate expression in today's mRNA vaccine platforms.
The development of prophylactic vaccines against Human Papillomavirus (HPV) represents a pivotal chapter in the history of virology and immunology, standing on the shoulders of centuries of innovation. From Edward Jenner's use of cowpox virus (a natural VLP of sorts) to confer protection against smallpox, through the advent of live-attenuated and inactivated whole-virus vaccines (e.g., polio, measles), to the modern era of subunit vaccines, the field has progressively sought safer and more targeted immunogens. The critical breakthrough for non-enveloped viruses like HPV was the shift from pathogen-based vaccines to antigen-based ones, specifically Virus-Like Particles (VLPs). VLPs are multimetric protein structures that mimic the native conformation of a virus but lack the viral genome, making them non-infectious and inherently safe. Their development for HPV, culminating in vaccines like Gardasil (Merck) and Cervarix (GSK), is a direct application of structural immunology—the rational design of vaccines based on the precise atomic-level understanding of antigen-antibody interactions and immune recognition.
Core Application Notes: HPV VLPs as Vaccine Antigens
Structural Basis: HPV is a non-enveloped, double-stranded DNA virus with an icosahedral capsid composed of two structural proteins: L1 (major) and L2 (minor). The immunodominant, type-specific neutralizing epitopes are located on the L1 protein. When expressed recombinantly (e.g., in yeast for Gardasil or insect cells for Cervarix), L1 proteins self-assemble into VLPs that are antigenically indistinguishable from the native virion.
Immune Recognition: The repetitive, high-density array of conformational epitopes on the VLP surface leads to:
- B-cell Activation: Efficient cross-linking of B-cell receptors, triggering potent T-cell-independent and T-cell-dependent humoral responses.
- Dendritic Cell Uptake: Enhanced uptake by follicular dendritic cells for superior antigen presentation.
- Memory Generation: Induction of long-lived plasma cells and memory B cells, providing durable protection.
Vaccine Formulations:
| Characteristic | Gardasil (Merck) | Gardasil9 (Merck) | Cervarix (GSK) |
|---|---|---|---|
| HPV Types | 6, 11, 16, 18 | 6, 11, 16, 18, 31, 33, 45, 52, 58 | 16, 18 |
| L1 Expression System | Saccharomyces cerevisiae (Yeast) | Saccharomyces cerevisiae (Yeast) | Trichoplusia ni insect cell line (Baculovirus) |
| Adjuvant | Amorphous Aluminum Hydroxyphosphate Sulfate (AAHS) | Amorphous Aluminum Hydroxyphosphate Sulfate (AAHS) | AS04 (Aluminum Hydroxide + MPL) |
| Key Efficacy Data | >98% protection vs. CIN2+ for types 16/18 | ~97% protection vs. high-grade lesions for 9 types | >90% sustained antibody titers at 10 years |
| Dosing Schedule | 0, 2, 6 months | 0, 2, 6 months | 0, 1, 6 months |
Detailed Experimental Protocols
Protocol 3.1: In Vitro Assembly and Purification of HPV L1 VLPs from Insect Cells
Objective: To produce and purify HPV-16 L1 VLPs using a baculovirus-insect cell expression system. Materials: Spodoptera frugiperda (Sf9) cells, recombinant baculovirus encoding HPV-16 L1, serum-free insect cell medium, lysis buffer (50 mM Tris-HCl pH 8.0, 0.5% Triton X-100, 400 mM NaCl, protease inhibitors), ultracentrifuge, CsCl or sucrose gradients. Procedure:
- Infection & Expression: Maintain Sf9 cells in suspension culture at 27°C. Infect at an MOI of 5-10 with recombinant baculovirus during mid-log phase.
- Harvest: 72 hours post-infection, pellet cells by centrifugation (500 x g, 10 min). Retain cell pellet.
- Lysis & Clarification: Resuspend cell pellet in ice-cold lysis buffer. Incubate on ice for 30 min with gentle agitation. Clarify lysate by centrifugation (10,000 x g, 30 min, 4°C).
- VLP Assembly & Purification: a. Salt Precipitation: Slowly add ammonium sulfate to the supernatant to 30% saturation. Incubate on ice for 1 hour. Pellet precipitated protein (10,000 x g, 20 min). b. Density Gradient Centrifugation: Resuspend pellet in PBS. Layer onto a pre-formed 20-60% (w/v) sucrose gradient. Ultracentrifuge at 150,000 x g for 3.5 hours at 4°C. c. Fraction Collection: Collect gradient fractions. Analyze fractions by SDS-PAGE and electron microscopy. Pool VLP-containing fractions.
- Buffer Exchange & Storage: Dialyze pooled fractions against PBS (pH 7.4). Concentrate using centrifugal filter units (100 kDa MWCO). Determine protein concentration, aliquot, and store at -80°C.
Protocol 3.2: Characterization of HPV VLPs by ELISA and Electron Microscopy
Objective: To assess the structural integrity and antigenic conformation of purified VLPs. Part A: Conformation-Specific ELISA Materials: Purified VLPs, neutralizing monoclonal antibody (e.g., H16.V5 for HPV-16), non-conformational antibody, 96-well high-binding plates, blocking buffer (5% non-fat milk in PBST), TMB substrate, plate reader. Procedure:
- Coating: Dilute VLPs to 1 µg/mL in PBS. Coat 96-well plate with 100 µL/well overnight at 4°C.
- Blocking: Aspirate and block with 200 µL/well blocking buffer for 1 hour at 37°C.
- Primary Antibody: Incubate with serial dilutions of conformational (H16.V5) and non-conformational antibodies in blocking buffer for 2 hours at 37°C.
- Detection: Wash plate (3x with PBST). Add appropriate HRP-conjugated secondary antibody for 1 hour at 37°C. Wash and develop with TMB. Stop reaction with 1M H₂SO₄. Read absorbance at 450 nm. Part B: Negative Stain Transmission Electron Microscopy (TEM) Materials: Purified VLP sample, 300-mesh carbon-coated copper grids, 2% uranyl acetate solution, TEM. Procedure:
- Grid Preparation: Glow-discharge grid to make it hydrophilic.
- Sample Application: Apply 5-10 µL of VLP sample to grid for 1 minute. Blot off excess with filter paper.
- Staining: Apply 5-10 µL of 2% uranyl acetate for 45 seconds. Blot off excess and air dry.
- Imaging: Examine grid under TEM at 80-100 kV. VLPs should appear as ~55 nm spherical particles with icosahedral symmetry.
Visualizations
Diagram Title: HPV VLP Assembly and B Cell Activation Pathway
Diagram Title: HPV VLP Production and Purification Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function in VLP Research | Example/Notes |
|---|---|---|
| Recombinant Baculovirus System | High-yield eukaryotic expression of L1 protein with proper folding and post-translational modifications. | Bac-to-Bac or flashBAC systems for insect cell (Sf9, Hi5) expression. |
| Saccharomyces cerevisiae Systems | Robust, scalable expression platform for L1 protein; used in licensed Gardasil production. | Commercially available yeast expression kits with optimized vectors and strains. |
| Conformation-Specific mAbs | Critical for ELISA-based quantification of properly assembled VLPs and neutralization assays. | H16.V5 (HPV-16), H18.J4 (HPV-18). Distinguish native from denatured capsids. |
| Sucrose or CsCl Gradients | Separation of fully assembled VLPs from capsomeres, aggregates, and host cell contaminants. | Pre-formed or self-forming gradients for ultracentrifugation-based purification. |
| Negative Stain EM Reagents | Rapid visualization of VLP morphology, size, and integrity. | Uranyl acetate (2%) or phosphotungstic acid for sample contrast. |
| Size-Exclusion Chromatography (SEC) | Analytical or preparative separation of VLPs based on hydrodynamic radius. | Superose 6 Increase columns for high-resolution analysis of assembly state. |
| AS04 & AAHS Adjuvants | Formulation components to enhance magnitude and duration of humoral immune response in vivo. | AS04 (Alum + MPL) induces strong Th1 response; AAHS is a proprietary aluminum salt. |
The history of viral vaccine development, from Edward Jenner's use of cowpox virus to confer protection against smallpox to the recent deployment of mRNA-LNP vaccines, is defined by the strategic harnessing of viral biology. A central pillar in this evolution is the development of viral vectors—viruses engineered to deliver foreign genetic material. This application note examines three critical platforms: Adenovirus (Ad, typically non-replicating), Vesicular Stomatitis Virus (VSV, replicating), and Modified Vaccinia Ankara (MVA, non-replicating). Their development represents a direct technological lineage from live-attenuated and inactivated vaccines, offering a balance between immunogenicity and safety tailored for modern prophylactic and therapeutic applications.
Platform Comparison: Characteristics & Quantitative Data
Table 1: Comparison of Viral Vector Platforms
| Feature | Adenovirus (Non-replicating, e.g., ChAdOx1) | Vesicular Stomatitis Virus (Replicating, e.g., VSV-ΔG) | Modified Vaccinia Ankara (Non-replicating) |
|---|---|---|---|
| Virus Family | Adenoviridae | Rhabdoviridae | Poxviridae |
| Genome Type | Linear dsDNA | Negative-sense ssRNA | Linear dsDNA |
| Packaging Capacity | ~8 kb (E1/E3 deleted) | ~4.5-5 kb (foreign gene insert) | >25 kb |
| Replication in Human Cells | No (E1 deleted) | Yes (cytoplasmic) | No (host-restricted) |
| Tropism | Broad (via fiber protein modification) | Broad (pantropic) | Broad (infects most mammalian cells) |
| Immunogenicity | High; strong humoral & cellular | Very High; robust innate & adaptive | High; potent Th1-biased cellular |
| Preexisting Immunity in Humans | High prevalence for common serotypes (e.g., Ad5) | Negligible | Low (smallpox vaccination ceased) |
| Key Safety Features | Non-replicating, low integration risk | Attenuated, oncolytic potential | Host-restricted, highly attenuated |
| Notable Licensed Use | COVID-19 vaccines (ChAdOx1-S, Ad26.COV2.S) | Ebola vaccine (ERVEBO) | Smallpox/Monkeypox vaccine (JYNNEOS) |
| Titer Achievable (Manufacturing) | ~10^11 – 10^12 VP/mL | ~10^8 – 10^9 PFU/mL | ~10^8 – 10^9 TCID50/mL |
Detailed Application Notes
Adenovirus Vectors (Non-replicating)
First-generation Adenovirus vectors, with deletions in early genes E1 and/or E3, are replication-incompetent in most cell lines. They transduce dividing and non-dividing cells efficiently, leading to high-level transgene expression. The major challenge is pre-existing immunity against common human serotypes, leading to vector neutralization. Strategies to circumvent this include using rare human serotypes (Ad26, Ad35) or non-human adenoviruses (ChAdOx1 from chimpanzee). Their role in rapid COVID-19 vaccine deployment underscored their utility as a pandemic-responsive platform.
Vesicular Stomatitis Virus (VSV) Vectors (Replicating)
VSV vectors are engineered by replacing the native glycoprotein (G) with a heterologous viral glycoprotein (e.g., Ebola virus GP). This creates a single-cycle, replication-competent vector that is highly immunogenic but attenuated. The VSV platform induces rapid and potent humoral immunity, as demonstrated by the ERVEBO vaccine. Its ability to replicate amplifies antigen load, but requires rigorous biosafety evaluation. Recent research explores its potent oncolytic activity against solid tumors.
Modified Vaccinia Ankara (MVA) Vectors (Non-replicating)
MVA is a highly attenuated poxvirus that underwent >500 passages in chicken embryo fibroblasts, resulting in the loss of ~15% of its genome and the ability to replicate productively in human and most mammalian cells. It infects cells and expresses early, intermediate, and late genes, but does not produce infectious progeny. This makes it exceptionally safe while maintaining strong immunogenicity, particularly for CD8+ T cell responses. It serves as a prime vaccine platform for diseases like HIV and malaria, and as a safer smallpox vaccine.
Experimental Protocols
Protocol 1: Titration of Non-replicating Adenovirus Vectors by TCID50 Assay Objective: Determine the infectious titer of an E1-deleted Adenovirus vector on HEK293 producer cells. Materials: HEK293 cell monolayer in 96-well plate, serially diluted virus stock, maintenance media (DMEM + 2% FBS), crystal violet stain. Procedure: 1. Seed HEK293 cells to achieve 80-90% confluency after 24 hours. 2. Prepare 10-fold serial dilutions of virus stock (10^-1 to 10^-10) in serum-free media. 3. Aspirate media from cell plate and inoculate 8 wells per dilution with 100 µL of diluted virus. 4. Include 8 control wells with media only. 5. Incubate at 37°C, 5% CO2 for 10-14 days. 6. Score wells for cytopathic effect (CPE) under a microscope. 7. Calculate titer using the Karber formula: TCID50/mL = 10^(1 + d - Σ(pi/n)), where d is log10 of the lowest dilution, pi is number of positive wells at dilution i, n is wells per dilution.
Protocol 2: In Vivo Immunogenicity Assessment of VSV-based Vaccine Objective: Evaluate humoral and cellular immune responses in mice post-immunization. Materials: 6-8 week old BALB/c mice, purified VSV-vectored vaccine, ELISA kit for antigen-specific IgG, IFN-γ ELISpot kit, flow cytometry reagents. Procedure: 1. Immunize mice (n=5/group) intramuscularly with 10^6 PFU of VSV vector in 50 µL PBS. 2. Collect serum samples at day 0 (pre-bleed), 14, and 28 post-immunization. 3. Perform endpoint ELISA on serum to quantify antigen-specific IgG titers. 4. At day 28, sacrifice mice and harvest spleens. 5. Prepare single-cell splenocyte suspension and perform IFN-γ ELISpot after 24-hour stimulation with relevant peptide pools. 6. For flow cytometry, stimulate splenocytes for 6 hours with peptides in the presence of brefeldin A, stain for surface markers (CD3, CD8, CD4) and intracellular cytokines (IFN-γ, TNF-α), and analyze.
Protocol 3: Generation of Recombinant MVA using Homologous Recombination Objective: Insert a foreign antigen gene into the MVA genome. Materials: MVA-BAC (bacterial artificial chromosome) genome, recombination plasmid with antigen flanked by MVA homology arms, Cre recombinase, permissive cells (e.g., BHK-21). Procedure: 1. Clone the antigen expression cassette (with a poxvirus promoter) into a transfer plasmid containing ~1 kb homology arms for a specific MVA deletion site (e.g., Del III). 2. Transfect the transfer plasmid into BHK-21 cells previously infected with wild-type MVA (MOI 0.05). 3. Harvest cells 48-72 hours post-transfection and lyse by freeze-thaw. 4. Screen for recombinant virus via multiple rounds of plaque purification under selective pressure (e.g., fluorescence or antibiotic selection). 5. Confirm genomic insertion by PCR and expression by western blot. 6. Amplify and purify recombinant MVA on chicken embryo fibroblast cells.
Visualizations
Title: Recombinant Adenovirus Production Workflow
Title: Immune Response Elicited by Viral Vectors
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Viral Vector Research
| Reagent | Function & Application | Example Vendor/Cat. No. (Representative) |
|---|---|---|
| HEK293 Cells | Producer cell line for E1-complemented Adenovirus propagation; high transfection efficiency. | ATCC CRL-1573 |
| BHK-21 Cells | Permissive cell line for production and titration of MVA and VSV vectors. | ATCC CCL-10 |
| Plaque Agarose Overlay | Semi-solid medium for viral plaque assay isolation and purification. | SeaPlaque Agarose (Lonza) |
| CsCl, UltraPure | Gradient medium for high-purity, research-scale ultracentrifugation of viral vectors. | Thermo Fisher, J61336.AP |
| Anti-Hexon Antibody (Adeno) | Detection of Adenovirus particles and quantification via ELISA or western blot. | Abcam, ab8250 |
| RNeasy Kit | RNA extraction from VSV-infected cells for viral replication or host response studies. | Qiagen, 74104 |
| IFN-γ ELISpot Kit | Quantification of antigen-specific T cell responses from immunized animal splenocytes. | Mabtech, 3321-2H |
| Fetal Bovine Serum (FBS), Charcoal/Dextran Stripped | Provides essential growth factors for cell culture while minimizing interference from hormones. | Gibco, 12676029 |
| Polyethylenimine (PEI), Linear | High-efficiency, low-cost transfection reagent for plasmid DNA into producer cells. | Polysciences, 23966 |
Within the historical continuum of viral vaccine development—from Jenner's empirical use of cowpox to the rational design of mRNA-LNP platforms—adjuvants have evolved from simple carriers to sophisticated immunomodulators. This progression mirrors the field's shift from whole-pathogen empiricism to molecular immunology. Early killed or subunit vaccines, while safer, often lacked potency, necessitating adjuvants to bridge innate and adaptive immunity. This document provides application notes and protocols for key modern adjuvants, contextualizing them as critical tools that enabled the transition from classical to contemporary vaccinology.
Application Notes & Quantitative Comparison
Table 1: Evolution and Characteristics of Key Adjuvant Platforms
| Adjuvant (Year Introduced) | Key Components | Proposed Primary Mechanism of Action | Key Licensed Vaccine Examples | Typical Cytokine Profile Induced |
|---|---|---|---|---|
| Alum (1930s) | Aluminum hydroxide or phosphate salts | NLRP3 inflammasome activation, depot formation, Th2 bias | Hepatitis B, DTaP, HPV | IL-4, IL-5, IL-13 (Th2); Weak Th1 |
| MF59 (1997) | Squalene-in-water nanoemulsion | Recruitment and activation of APCs at injection site, enhanced antigen uptake | Enhanced influenza (Fluad) | Robust IgG, Th1/Th2 balanced, T FH |
| AS01 (2009) | QS-21 + MPL + Liposome | MPL (TLR4 agonist) and QS-21 synergize, strong innate stimulation | Shingles (Shingrix), Malaria (RTS,S) | Strong IFN-γ, IL-2 (Th1), CD8+ T cells |
| CpG 1018 (2017) | Class B CpG ODN (TLR9 agonist) | B cell and pDC activation via TLR9, Th1 bias | Hepatitis B (Heplisav-B) | IFN-α, IL-6, IFN-γ (Th1) |
| mRNA-LNP (2020s) | Ionizable lipid, PEG-lipid, cholesterol, phospholipid | In vivo transfection, innate sensing via TLRs/RLRs, self-adjuvanting | SARS-CoV-2 (Moderna, Pfizer-BioNTech) | Type I IFN, Th1, strong T FH & CD8+ |
Table 2: Experimental Readouts for Adjuvant Comparison In Vivo (Mouse Model)
| Parameter | Alum | MF59 | AS01 | CpG ODN | Measurement Protocol (Summary) |
|---|---|---|---|---|---|
| Antigen-Specific IgG Titer (GMT) | High (Th2-isotypes) | Very High | Exceptional High | High | ELISA on serum, endpoint dilution. |
| IgG2c/IgG1 Ratio | Low (~0.1) | Moderate (~1-2) | High (>3) | High (>5) | Isotype-specific ELISA. Ratio indicates Th1 bias. |
| IFN-γ+ CD4+ T cells (Spot Forming Units) | Low (<50) | Moderate (200-500) | High (1000-3000) | High (800-2500) | ELISpot on splenocytes re-stimulated with antigen. |
| Germinal Center B Cells (Frequency %) | Low-Mod (1-2%) | High (3-5%) | Very High (5-8%) | High (3-6%) | Flow cytometry (B220+ CD95+ GL7+) from draining LN/spleen. |
| Antigen Depot Duration | Long (2-3 weeks) | Short (<24h) | Short (<24h) | Short (<24h) | In vivo imaging with fluorescent/radiolabeled antigen. |
Experimental Protocols
Protocol 1: Formulation of a Model Subunit Vaccine with AS01-like Adjuvant
Objective: To prepare a liposomal adjuvant formulation containing MPLA and QS-21 for co-administration with a recombinant protein antigen. Materials: See "Scientist's Toolkit" below. Procedure:
- Liposome Preparation: Dissolve DOPC and cholesterol (55:45 molar ratio) in chloroform in a round-bottom flask. Remove solvent under rotary evaporation to form a thin lipid film. Hydrate the film with sterile PBS (pH 7.4) at 60°C for 1h with gentle agitation to form multilamellar vesicles (MLVs).
- Size Reduction & MPLA Incorporation: Extrude the MLV suspension through a 100nm polycarbonate membrane filter (Avanti Mini-Extruder) 21 times. Add MPLA (aqueous suspension) to the pre-formed liposomes and incubate at 25°C for 30 min with stirring.
- QS-21 Addition: Add purified QS-21 saponin to the MPLA-liposome mixture to achieve final target concentrations (e.g., 50 µg/mL MPLA, 50 µg/mL QS-21). Incubate for 15 min at 25°C.
- Antigen Mixing: Combine the adjuvant formulation with the recombinant protein antigen (e.g., SARS-CoV-2 RBD) at the desired ratio (e.g., 1:1 v/v) just prior to immunization. Do not freeze-thaw.
- QC: Determine particle size and PDI by dynamic light scattering (DLS) and zeta potential by electrophoretic light scattering. Endotoxin level should be <1 EU/mL (LAL test).
Protocol 2: Evaluating Adjuvant-Induced Innate Immunity in a Murine Model
Objective: To profile early cytokine and cellular responses in the draining lymph node (dLN) post-immunization. Procedure:
- Immunization: C57BL/6 mice (n=5/group) receive 50µL intramuscular injection of antigen (10µg) formulated with test adjuvant or control (PBS/Alum). Use both hind legs for sufficient cell yield.
- dLN Harvest & Processing: Euthanize mice at 6h (cytokines) and 18-24h (cell phenotyping). Excise popliteal and inguinal LNs. Mechanically dissociate through a 70µm cell strainer to create single-cell suspensions.
- Cytokine Quantification (6h): Homogenize dLN tissue in PBS with protease inhibitors. Clarify supernatant by centrifugation. Analyze levels of IL-6, TNF-α, IFN-γ, and IL-12p70 using a multiplex bead-based assay (e.g., Luminex) or ELISA.
- Flow Cytometry (18h): Stain single-cell suspension with viability dye and antibodies: CD11b, CD11c, MHC-II, Ly6C, Ly6G. Identify recruited monocytes (CD11b+ Ly6Chi), neutrophils (CD11b+ Ly6Cmid Ly6G+), and activated dendritic cells (CD11c+ MHC-IIhi). Acquire data on a flow cytometer and analyze using FlowJo software.
- Statistical Analysis: Use one-way ANOVA with Tukey's post-hoc test for multiple comparisons. P < 0.05 is considered significant.
Signaling Pathways & Workflow Visualizations
Title: Adjuvant Mechanisms from Innate Sensing to T Cell Activation
Title: In Vivo Adjuvant Evaluation Workflow Timeline
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Adjuvant Formulation and Testing
| Item | Supplier Examples | Function in Protocol |
|---|---|---|
| Monophosphoryl Lipid A (MPLA, Synthetic) | InvivoGen, Sigma-Aldrich | TLR4 agonist component of AS01; activates APCs via TRIF/MyD88. |
| QS-21 Saponin (Purified) | Desert King International, InvivoGen | Saponin component of AS01; enhances cellular responses, synergizes with MPLA. |
| DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) | Avanti Polar Lipids, Sigma-Aldrich | Primary phospholipid for forming stable, neutral liposomes. |
| Squalene (for MF59-like emulsions) | Sigma-Aldrich, Croda | Biocompatible oil phase of nanoemulsion adjuvants. |
| CpG ODN 1826 (Class B) / 1018 (Human) | InvivoGen, Integrated DNA Technologies | TLR9 agonist; induces strong Th1 and B cell responses. |
| Aluminum Hydroxide Gel (2% Alhydrogel) | InvivoGen, Brenntag | Standard Th2 adjuvant control for in vivo studies. |
| Polycarbonate Membrane Extruder (100nm) | Avanti Polar Lipids | For generating uniform, small unilamellar liposomes. |
| Luminex Multiplex Mouse Cytokine Panel | Thermo Fisher, R&D Systems | Simultaneously quantifies multiple cytokines from small sample volumes. |
| Fluorochrome-conjugated Antibodies (CD11b, CD11c, MHC-II, B220, GL7, CD95) | BioLegend, BD Biosciences | For flow cytometry analysis of innate cell recruitment and GC reactions. |
| Mouse IFN-γ ELISpot Kit | Mabtech, BD Biosciences | Quantifies antigen-specific T cell responses at single-cell level. |
Application Notes & Protocols
The journey from Edward Jenner's empirical use of cowpox to prevent smallpox to the rational design of mRNA vaccines represents a paradigm shift in vaccinology. Classical methods relied on pathogen cultivation and attenuation. The late 20th century introduced reverse vaccinology, pioneered for Neisseria meningitidis B, which uses genomic data to identify antigens without culturing the pathogen. This approach, combined with structural biology and computational design, has revolutionized antigen selection and optimization, as exemplified by the development of prefusion-stabilized respiratory syncytial virus (RSV) F protein vaccines.
Genomic Mining for Antigen Identification
Objective: To computationally identify and prioritize RSV surface-exposed proteins as potential vaccine candidates from its genomic sequence.
Protocol 1.1: In Silico Antigen Screening Workflow
- Data Acquisition: Download the complete genomic sequence of RSV A2 strain (NCBI Reference Sequence: NC_038235.1) using tools like
efetchfrom the Entrez Direct utilities. - Open Reading Frame (ORF) Prediction: Use prediction software (e.g., GeneMarkS, Glimmer) to identify all potential protein-coding genes.
- Subcellular Localization Prediction: Submit predicted protein sequences to algorithms (e.g., PSORTb, SignalP, TMHMM) to identify proteins with signal peptides, transmembrane domains, or outer membrane anchoring motifs, indicating surface exposure.
- Conservation Analysis: Perform a BLASTP search of candidate antigens against a database of diverse RSV strains (A, B subgroups) to calculate percent sequence identity. Prioritize highly conserved antigens.
- Antigenicity & Epitope Prediction: Analyze candidates for predicted B-cell and T-cell epitopes using tools like BepiPred-2.0 and NetMHCIIpan.
Table 1: Computational Screening Results for Key RSV Antigens
| Antigen | Gene Length (bp) | Predicted Localization | % Conservation (Across A & B strains) | Predicted B-cell Epitope Count | Priority Rank |
|---|---|---|---|---|---|
| Fusion (F) Glycoprotein | 1902 | Surface (Type I TM) | 99% | 12 | 1 |
| Glycoprotein (G) | 945 | Surface (Type II TM) | 53% (Variable) | 8 | 3 |
| Small Hydrophobic (SH) Protein | 393 | Surface (Oligomeric) | 85% | 3 | 4 |
| Attachment (G) Glycoprotein* | 1782 | Surface (Type II TM) | 92% | 10 | 2 |
Note: The G protein is highly glycosylated; sequence variability is a key challenge. The F protein emerges as the top candidate due to high conservation and essential role in viral entry.
Structural Vaccinology: Stabilizing the Prefusion F Conformation
Objective: Engineer the metastable prefusion conformation of the RSV F protein to present neutralization-sensitive epitopes.
Background: The RSV F protein undergoes a major conformational change from prefusion (pre-F) to postfusion (post-F) states. Neutralizing antibodies primarily target the pre-F state. The post-F state is more stable but immunogenically suboptimal.
Protocol 2.1: Structure-Based Antigen Design
- Structural Analysis: Obtain atomic coordinates for pre-F (e.g., PDB: 4MMQ) and post-F (e.g., PDB: 3RRR) conformations. Superimpose structures using PyMOL or ChimeraX to identify regions of high conformational variance (e.g., F-perfusion-specific antigenic site Ø, located at the membrane-distal apex).
- Cavity-Filling Mutations: Identify cavities in the pre-F trimer interface unique to this state. Introduce cavity-filling mutations (e.g., S190F, V207L) to increase hydrophobic packing and stabilize the trimer.
- Disulfide Bond Engineering: Introduce a pair of cysteine residues (e.g., S155C & S290C) to form a covalent "tether" (disulfide bond) that physically prevents the transition to the post-F conformation. Validate bond formation via non-reducing SDS-PAGE.
- Foldon Trimerization Domain: Replace the native viral transmembrane and cytoplasmic domains with a synthetic foldon trimerization motif (from T4 bacteriophage fibritin) to ensure homogeneous, soluble trimer expression in mammalian cell systems (e.g., HEK293F).
Key Research Reagent Solutions
| Reagent / Material | Function / Rationale |
|---|---|
| pCAGGS Expression Vector | Mammalian expression vector with strong chicken beta-actin promoter for high-yield transient protein production. |
| Expi293F Cells | A clonal derivative of HEK293 cells adapted for high-density suspension culture and high transient protein yield. |
| Freestyle 293 Expression Medium | Serum-free medium optimized for Expi293F cell growth and transfection. |
| PEI MAX 40K (Polyethylenimine) | High-efficiency, low-cost cationic polymer for transient plasmid DNA transfection. |
| Strep-tactin XT 4Flow resin | Affinity chromatography resin for gentle purification of Strep-tag II-fused F protein variants. |
| DS-Cav1 (F protein mutant) | Reference prefusion-stabilized immunogen containing cavity-filling mutations (S190F, V207L) and an engineered disulfide bond (S155C, S290C). |
Protocol 2.2: Expression & Purification of Engineered pre-F Antigen
- Transfection: Cultivate Expi293F cells at 37°C, 8% CO₂, 125 rpm to a density of 3.0 x 10⁶ cells/mL. Transfect using PEI MAX (1:3 DNA:PEI ratio) with plasmid encoding the engineered F construct (e.g., DS-Cav1 with C-terminal foldon and affinity tag).
- Harvest: 5-7 days post-transfection, centrifuge culture at 4,000 x g for 30 min. Filter supernatant through a 0.22 µm PES membrane.
- Affinity Chromatography: Load filtered supernatant onto a Strep-tactin XT column equilibrated with Buffer W (100 mM Tris, 150 mM NaCl, pH 8.0). Wash with 10 column volumes (CV) of Buffer W. Elute with 5 CV of Buffer E (Buffer W + 50 mM biotin).
- Size-Exclusion Chromatography (SEC): Concentrate eluate and inject onto a Superose 6 Increase 10/300 GL column pre-equilibrated in formulation buffer (e.g., PBS, pH 7.4). Collect the peak corresponding to the trimeric fraction.
- Quality Control: Analyze SEC peak by SDS-PAGE (reducing/non-reducing), negative-stain Electron Microscopy, and Differential Scanning Calorimetry (DSC) to confirm purity, oligomeric state, and thermal stability (Tm).
Table 2: Biophysical Characterization of RSV F Protein Constructs
| Construct | Conformation | SEC Elution Volume (mL) | Apparent Molecular Weight (kDa) | Melting Temperature Tm (°C) | Neutralizing Antibody Titer (Murine Model, Log10) |
|---|---|---|---|---|---|
| Wild-type Soluble F | Pre/Post Mix | 13.5 | ~600 (heterogeneous) | 44.7, 58.3 (two peaks) | 3.2 |
| Post-F (Stable) | Postfusion | 14.8 | ~540 | 68.5 | 2.8 |
| DS-Cav1 | Prefusion-stabilized | 12.1 | ~620 (homogeneous trimer) | 65.1 (single peak) | 5.1 |
Immunogenicity Assessment Protocol
Protocol 3.1: Murine Model Immunization & Analysis
- Immunization: Groups (n=10) of BALB/c mice (6-8 weeks old) receive 10 µg of antigen (e.g., DS-Cav1, post-F, buffer control) formulated with 25% (v/v) AddaVax (MF59-like adjuvant) via intramuscular injection at weeks 0 and 4.
- Serum Collection: Collect blood via retro-orbital bleed at week 6. Isolate serum by centrifugation (10,000 x g, 10 min).
- ELISA for Binding Antibodies: Coat high-binding plates with pre-F or post-F protein (2 µg/mL). Perform serial dilution of serum. Detect bound IgG using HRP-conjugated anti-mouse IgG and TMB substrate. Report endpoint titers.
- Microneutralization Assay: Incubate RSV A2 stock (100 TCID₅₀) with serial serum dilutions for 1h. Add mixture to HEp-2 cell monolayers in 96-well plates. After 5 days, fix cells and immunostain for RSV nucleoprotein. Neutralization titer (NT₅₀) is the serum dilution causing 50% reduction in infected foci.
Visualization Diagrams
Title: Reverse Vaccinology and Structural Design Workflow
Title: RSV F Protein Conformational States & Immune Recognition
Overcoming Hurdles: Stability, Immunogenicity, and Manufacturing Challenges Across Platforms
Application Notes
The development of live-attenuated vaccines (LAVs) represents a cornerstone achievement in the history of virology, from Jenner’s empirical use of cowpox to the rational attenuation of viruses like polio. For poliovirus type 2, this balance is critical. The global cessation of oral polio vaccine (OPV) type 2 use in 2016 due to circulating vaccine-derived poliovirus (cVDPV) emergence underscores the reversion risk. Current strategies focus on novel OPV2 (nOPV2), genetically stabilized to reduce reversion while maintaining immunogenicity.
Table 1: Comparison of Historical and Novel Poliovirus Type 2 Vaccine Strains
| Parameter | Sabin OPV2 (Historical) | Novel OPV2 (nOPV2) | Measurement/Notes |
|---|---|---|---|
| Genetic Stability (Reversions) | High (cVDPV2 frequency) | Significantly Reduced | Measured as cVDPV2 cases post-switch; nOPV2 shows >80% reduction in emergences. |
| Seroconversion Rate (after 1 dose) | >90% (in infants) | >90% (in infants) | Neutralizing antibody titer ≥1:8. |
| Mean Virus Shedding Duration | ~4-6 weeks | ~3-5 weeks | Duration of fecal shedding post-vaccination. |
| Key Genomic Modifications | 3 attenuation loci (5' UTR, VP1) | 3 attenuation loci + 2 stabilizers (ribosome entry site, polymerase fidelity) | Designed to prevent recombination and reversion. |
| Temperature Sensitivity | Moderate (loss at >37°C) | Enhanced | Reduced risk of reversion at higher temperatures. |
| Regulatory Status | WHO Prequalification (1988) | WHO EUL (2020), Prequalified (2023) | nOPV2 is the first vaccine under WHO’s Emergency Use Listing. |
Table 2: Key Outcomes from nOPV2 Clinical Trials (Pooled Data)
| Trial Phase | Participants (n) | Seroconversion Rate (%) | Vaccine Virus Shedding at Day 7 (%) | cVDPV2 Cases Linked to Trial | Follow-up Period |
|---|---|---|---|---|---|
| Phase II | ~400 | 92.5 | 21.3 | 0 | 6 months |
| Phase III | ~1500 | 94.1 | 19.8 | 0 | 12 months |
| Real-World Use (2021-2023) | ~600 million doses | N/A | Surveillance-based | <5* | Ongoing |
*Reported as limited, genetically-linked emergences with minimal neurovirulence.
Experimental Protocols
Protocol 1:In VitroAssessment of Genetic Stability and Reversion Risk
Objective: To quantify the mutation rate and reversion frequency of nOPV2 compared to Sabin OPV2 through serial passage.
Materials:
- RD (rhabdomyosarcoma) or HEp-2C cell lines.
- Viral stocks: Sabin OPV2 and nOPV2.
- Cell culture media (DMEM + 2% FBS).
- RT-PCR and sequencing reagents.
- Plaque assay materials (agar overlay, staining).
Methodology:
- Serial Passage: Infect cell monolayers at a low MOI (0.1). Harvest virus when full CPE is observed (3-4 days). Use supernatant to infect fresh cells. Repeat for 30 passages.
- Plaque Phenotype Analysis: At passages 0, 10, 20, and 30, perform plaque assays under semi-solid overlay at 34.5°C (permissive) and 39.5°C (restrictive). Calculate the ratio of plaque-forming units (pfu) at 39.5°C/34.5°C as a measure of temperature-sensitive (ts) phenotype reversion.
- Genetic Sequencing: Extract viral RNA from harvested passages. Perform RT-PCR amplifying key genomic regions (5' UTR, VP1, 3D polymerase). Use next-generation sequencing (NGS) for a minimum depth of 10,000x to identify minority variants and recombination events.
- Data Analysis: Calculate mutation frequency per passage. Identify specific reversions at known attenuation sites (e.g., nucleotide 481 in 5' UTR, amino acid 143 in VP1).
Protocol 2:In VivoImmunogenicity and Shedding Study in Transgenic Mouse Model
Objective: To compare the immunogenicity and duration of shedding of nOPV2 versus Sabin OPV2 in a susceptible animal model.
Materials:
- TgPVR21 transgenic mice (express human poliovirus receptor).
- Viral inoculum (10^5 pfu/dose in 50µL).
- Serum collection tubes.
- Fecal sample collection apparatus.
- Plaque assay or qRT-PCR for virus quantification.
- Microneutralization assay reagents.
Methodology:
- Immunization: Randomly assign mice (n=20 per group) to receive a single intramuscular (IM) injection of either nOPV2, Sabin OPV2, or placebo.
- Sample Collection:
- Shedding: Collect fecal pellets individually on days 1, 3, 5, 7, 10, and 14 post-inoculation. Homogenize and titrate for infectious virus via plaque assay.
- Immunogenicity: Collect serum pre-inoculation and on day 28. Inactivate at 56°C for 30 minutes.
- Microneutralization Assay: Perform a standard neutralization assay using Sabin 2 reference strain. Seroconversion is defined as a neutralizing antibody titer ≥1:8 in previously seronegative animals.
- Statistical Analysis: Compare mean log10 virus titers in feces over time (longitudinal mixed model). Compare geometric mean titers (GMTs) and seroconversion rates between groups (ANOVA, chi-square).
Visualizations
Title: Assessing Genetic Stability of OPV2 Strains
Title: LAV Immunogenicity vs Reversion Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for OPV2 Research
| Item | Function & Application | Example/Notes |
|---|---|---|
| RD or HEp-2C Cell Line | Permissive for poliovirus replication; used for virus propagation, titration (plaque assay), and neutralization tests. | ATCC CCL-136 (RD cells). |
| TgPVR21 Transgenic Mice | Express human poliovirus receptor; essential in vivo model for neurovirulence, immunogenicity, and shedding studies. | Available from specific repositories under MTA. |
| Poliovirus Neutralizing Antibody Reference Serum | Standard for calibrating microneutralization assays to ensure inter-lab comparability of immunogenicity data. | WHO International Standard (Type 2, code 66/204). |
| nOPV2 & Sabin OPV2 Reference Reagents | Genetically characterized master virus seeds for use as controls in all assays. | Obtained from WHO or CDC repositories. |
| Next-Generation Sequencing (NGS) Kit | For deep sequencing of viral RNA to identify low-frequency reversions and recombinants. | e.g., Illumina COVIDSeq Test (adapted for poliovirus). |
| Plaque Assay Agar Overlay | Semi-solid medium to restrict viral spread, allowing visualization and counting of discrete plaques for virus quantification. | 1-2% Agarose or Avicel in maintenance media. |
| Real-Time RT-PCR Assay for Poliovirus | Rapid, sensitive detection and quantification of poliovirus RNA in clinical (stool) or research samples. | WHO-recommended primers/probes targeting VP1 region. |
| Temperature-Controlled Incubators | Critical for assessing temperature-sensitive (ts) phenotype, a key marker of attenuation stability. | Requires precise calibration at 34.5°C, 37.0°C, and 39.5°C. |
Application Notes
Historical Context and Modern Challenge
Within the arc of vaccine development from Jenner's empiricism to mRNA precision, inactivated and subunit vaccines represent a critical middle chapter, offering superior safety over live-attenuated platforms but often at the cost of immunogenicity. Unlike whole-pathogen vaccines, these platforms lack the inherent danger signals and antigenic breadth needed for robust innate immune activation and cytotoxic T-cell priming, which are essential for combating intracellular pathogens and achieving durable immunity. Modern research focuses on rational adjuvant design and delivery systems to overcome these limitations, bridging the historical safety goals of inactivation with the contemporary need for potent cellular immunity.
Key Immunological Hurdles and Quantitative Benchmarks
The primary challenges are quantitatively demonstrable in comparative immunogenicity studies.
Table 1: Comparative Immunogenicity Profiles of Vaccine Platforms
| Platform | Typical Adjuvant Used | Mean IgG Titer (Log10) | CD8+ T-cell Response (IFN-γ SFU/10^6 cells) | Key Limitation |
|---|---|---|---|---|
| Live-Attenuated | None (self-adjuvanting) | 4.5 - 5.5 | 800 - 1500 | Reactogenicity |
| Inactivated (Whole Virion) | Alum | 3.8 - 4.5 | 50 - 200 | Weak CTL priming |
| Protein Subunit | Alum | 3.5 - 4.2 | 10 - 100 | Poor Th1/CTL response |
| Protein Subunit | AS01/AS04 | 4.2 - 5.0 | 300 - 700 | Cost, complexity |
| mRNA-LNP | LNP (ionizable lipid) | 5.0 - 6.0+ | 1000 - 2500+ | Stability, reactogenicity |
Table 2: Impact of Advanced Adjuvant Systems on Subunit Vaccine Responses
| Adjuvant System (Example) | TLR Agonist Included | Target PRR | Fold-Improvement vs. Alum (IgG) | Fold-Improvement vs. Alum (CD8+ T-cells) |
|---|---|---|---|---|
| AS01 (Shingrix) | QS-21 (saponin) + MPL | TLR4 | 8-12x | 20-50x |
| AS04 (Cervarix) | MPL | TLR4 | 3-5x | 5-10x |
| CpG 1018 + Alum (Heplisav-B) | CpG ODN | TLR9 | 10-15x | 15-30x |
| MF59 (Fluad) | None (squalene emulsion) | N/A | 2-4x | 1.5-3x |
Core Strategies for Enhancement
- Adjuvant Innovation: Moving from aluminum salts (Alum), which predominantly elicit Th2-biased antibody responses, to Toll-like Receptor (TLR) agonists (e.g., MPL (TLR4), CpG (TLR9)) that directly activate dendritic cells (DCs) to promote Th1 and cytotoxic T-lymphocyte (CTL) responses.
- Delivery Technologies: Employing nanoparticle carriers (e.g., liposomes, virus-like particles (VLPs), polymeric nanoparticles) to promote antigen drainage to lymph nodes, enhance DC uptake, and enable co-delivery of antigen and adjuvant to the same APC.
- Rational Antigen Design: Using structural biology to engineer stabilized immunogens (e.g., prefusion F protein for RSV) that expose key neutralizing epitopes, improving antibody quality.
Detailed Protocols
Protocol 1: Evaluating T-cell Priming by ELISpot
Objective: Quantify antigen-specific CD8+ T-cell responses (IFN-γ secretion) from splenocytes of vaccinated mice.
Materials:
- Mouse IFN-γ ELISpot kit (e.g., Mabtech)
- Multiscreen HTS IP filter plates (Millipore)
- Concanavalin A (positive control)
- RPMI-1640 complete media
- Peptide pools spanning target antigen
- ELISpot plate reader
Procedure:
- Splenocyte Isolation: Euthanize mouse 14 days post-boost. Aseptically remove spleen.
- Homogenize spleen through a 70 µm cell strainer into complete RPMI. Lyse red blood cells using ACK buffer.
- Wash cells twice, count, and resuspend at 5 x 10^6 cells/mL.
- ELISpot Plate Setup: Coat ELISpot plate with anti-mouse IFN-γ capture antibody (15 µg/mL in PBS) overnight at 4°C.
- Block plate with complete RPMI for 2 hours at 37°C.
- Add 100 µL cell suspension (5 x 10^5 cells/well) to triplicate wells.
- Add 100 µL of peptide pool (final conc. 2 µg/mL/peptide), media (negative control), or ConA (positive control).
- Incubate plate for 40 hours at 37°C, 5% CO2.
- Develop plate per kit instructions: incubate with detection antibody, then streptavidin-enzyme conjugate, followed by chromogenic substrate.
- Analysis: Air-dry plate and count spots using an automated ELISpot reader. Report as mean Spot-Forming Units (SFU) per 10^6 cells ± SEM.
Protocol 2: Formulation of Antigen-Adjuvant Nanoparticles
Objective: Co-encapsulate subunit antigen and TLR agonist in biodegradable PLGA nanoparticles.
Materials:
- PLGA (50:50, acid-terminated, MW 10-15 kDa)
- Recombinant antigen protein
- TLR agonist (e.g., Resiquimod (R848) for TLR7/8)
- Polyvinyl alcohol (PVA)
- Dichloromethane (DCM)
- Probe sonicator
Procedure:
- Oil Phase: Dissolve 50 mg PLGA and 0.5 mg TLR agonist in 2 mL DCM.
- Aqueous Phase: Dissolve 1 mg antigen in 0.5 mL of 2% (w/v) PVA solution.
- Emulsification: Add the aqueous phase to the oil phase. Emulsify using a probe sonicator (30% amplitude, 60 seconds on ice) to form a primary water-in-oil (W/O) emulsion.
- Double Emulsion: Add this primary emulsion to 4 mL of 2% PVA solution and sonicate again (30% amplitude, 90 seconds) to form a stable water-in-oil-in-water (W/O/W) emulsion.
- Solvent Evaporation: Stir the double emulsion at room temperature for 4 hours to evaporate DCM.
- Harvesting: Centrifuge nanoparticles at 20,000 x g for 30 minutes at 4°C. Wash pellet 3x with sterile water to remove PVA and unencapsulated material.
- Characterization: Resuspend in PBS. Measure particle size and PDI by DLS, zeta potential by electrophoresis, and antigen/adjuvant loading efficiency via BCA assay and HPLC, respectively.
Visualizations
Title: Adjuvant-Driven Immune Enhancement Pathway
Title: Nanoparticle Vaccine Workflow & Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Enhanced Inactivated/Subunit Vaccine Research
| Reagent Category | Specific Example | Function & Rationale |
|---|---|---|
| Pattern Recognition Receptor (PRR) Agonists | Monophosphoryl Lipid A (MPL, TLR4), CpG ODN 1018 (TLR9), Resiquimod/R848 (TLR7/8) | Provides "danger signal" to innate immunity, promotes DC maturation, Th1 bias, and cross-presentation essential for CTL priming. |
| Nanoparticle Delivery Systems | Poly(lactic-co-glycolic acid) (PLGA), Liposomes (DOPC, Cholesterol), Cationic polymers (e.g., PEI) | Protects antigen, enables lymph node targeting, facilitates co-delivery of antigen and adjuvant to same APC, and enhances antigen persistence. |
| Characterization Assays | Dynamic Light Scattering (DLS), Bicinchoninic Acid (BCA) Assay, IFN-γ ELISpot Kits, Multiplex Cytokine Panels | Measures nanoparticle size/PDI, quantifies protein antigen loading, and evaluates critical cellular immune responses (CTL, Th1). |
| Adjuvant Formulations | Adju-Phos (Aluminum phosphate), MF59 (squalene emulsion), ISCOMATRIX (saponin-based) | Classic (Alum) and modern comparators for benchmarking novel formulations for humoral vs. cellular immunity. |
| T-cell Assay Reagents | Fluorescent MHC class I tetramers, intracellular staining antibodies (anti-IFN-γ, anti-TNF-α, anti-IL-2), cell proliferation dyes (CFSE) | Enables precise quantification and phenotypic characterization of antigen-specific T-cell populations by flow cytometry. |
Application Notes & Protocols
Thesis Context: Within the historical arc of viral vaccine development—from Jenner’s empirical use of cowpox to the rational design of mRNA vaccines—the reliance on continuous refrigeration (the "cold chain") has been a critical, yet fragile, cornerstone. This dependency is a direct consequence of the inherent thermodynamic instability of traditional biologicals, including live-attenuated, inactivated, and protein-subunit vaccines. The emergence of mRNA-LNP platforms, with their own distinct stability profiles, represents a potential paradigm shift, yet the logistical challenges of thermal stability remain paramount for global distribution.
Quantitative Stability Data of Representative Viral Vaccines
Table 1: Thermal Degradation Profiles of Traditional Biological Vaccines vs. mRNA-LNP Platform
| Vaccine Platform (Example) | Stable Long-Term Storage | Real-Time Stability at 2-8°C (Months) | Accelerated Degradation at 25°C | Key Instability Factor(s) |
|---|---|---|---|---|
| Live-Attenuated (MMR II) | ≤ -70°C (lyophilized) | 24 | Loses potency within days/weeks | Protein denaturation, viral particle aggregation, loss of replicative fidelity. |
| Inactivated Split-Virion (Influenza) | 2-8°C | 12-18 | Potency drop >1 log in weeks | HA protein aggregation, dissociation of viral components. |
| Protein Subunit (Recombinant Hepatitis B) | 2-8°C | 24-36 | Particle aggregation over months | Protein unfolding, surface adsorption, covalent dimerization. |
| Viral Vector (Adenovirus-based, e.g., Ebola) | ≤ -60°C (often) | ≤ 6 (varies) | Rapid loss of infectivity titer | Vector aggregation, protein capsid degradation, DNA integrity loss. |
| mRNA-LNP (COVID-19 vaccines) | -70°C to -20°C (initial) | 3-9* (post-thaw) | Lipid nanoparticle fusion, mRNA hydrolysis | mRNA chemical degradation (hydrolysis), LNP physical instability (fusion, size change), freeze-thaw stress. |
Note: Modern formulations aim for extended 2-8°C stability (up to 18 months for some). Data synthesized from FDA labels, WHO PIS documents, and recent peer-reviewed stability studies.
Experimental Protocols for Stability Assessment
Protocol 2.1: Accelerated Stability Study for Vaccine Antigenicity
Objective: To predict the real-time stability of a protein-subunit or inactivated viral vaccine at recommended storage (2-8°C) using elevated temperatures.
Materials:
- Vaccine samples (filled in final container closure).
- Stability chambers set at 5°C (control), 25°C, and 37°C.
- ELISA kit specific for vaccine antigen (e.g., anti-HA for influenza).
- Dynamic Light Scattering (DLS) instrument.
- SDS-PAGE and CE-SDS equipment.
Methodology:
- Sample Allocation: Place identical vaccine vials/lots into controlled stability chambers at 5°C ± 3°C, 25°C ± 2°C, and 37°C ± 2°C.
- Time Points: Remove samples in triplicate at t=0, 1, 2, 4, 8, and 12 weeks.
- Antigenicity Assay (ELISA):
- Coat ELISA plates with a capture antibody specific for the native conformational epitope of the vaccine antigen.
- Apply serially diluted stability samples and a reference standard.
- Detect using a labeled secondary antibody. Plot relative potency vs. control.
- Physical Stability (DLS):
- Dilute sample as per manufacturer's guideline in formulation buffer.
- Measure hydrodynamic diameter (Z-average) and polydispersity index (PDI).
- A >10% increase in size or PDI >0.2 indicates particle aggregation.
- Chemical Stability (CE-SDS):
- Perform reduced and non-reduced capillary electrophoresis SDS.
- Monitor increases in fragment peaks or aggregates relative to main product peak.
Data Interpretation: Use the Arrhenius equation (k = A e^(-Ea/RT)) to model degradation rate constants (k) at higher temperatures and extrapolate to 5°C, estimating the time to a pre-defined potency limit (e.g., 95% of label claim).
Protocol 2.2: Forced Degradation of mRNA-LNP Formulations
Objective: To identify primary degradation pathways in mRNA-LNP vaccines under thermal and mechanical stress.
Materials:
- mRNA-LNP drug product.
- Ribogreen fluorescence assay kit.
- Particle size analyzer (NTA or DLS).
- HPLC system with ion-pair reversed-phase column.
- Freeze-thaw cycler or -80°C freezer/water bath.
Methodology:
- Thermal Stress: Incubate LNP samples at 4°C, 25°C, and 40°C for 0, 1, 3, and 7 days.
- Freeze-Thaw Stress: Subject samples to 1, 3, and 5 cycles of freezing (-80°C for 1 hr) and thawing (25°C water bath).
- mRNA Integrity Analysis (IP-RP HPLC):
- Extract mRNA from LNPs using a destabilizing buffer (e.g., containing Triton X-100).
- Inject onto HPLC. Monitor for main peak (full-length mRNA) and earlier eluting peaks (degraded fragments).
- Encapsulation Efficiency (Ribogreen Assay):
- Measure total mRNA (sample lysed with detergent) vs. unencapsulated mRNA (sample in buffer).
- Calculate % encapsulated = (1 - (unencapsulated/total)) x 100. A drop indicates LNP membrane disruption.
- Particle Characterization (DLS/NTA):
- Measure particle size, PDI, and concentration pre- and post-stress.
- Significant size increase indicates aggregation/fusion; decrease indicates disintegration.
Visualizations (Graphviz DOT)
Title: Vaccine Platform Degradation Pathways (82 chars)
Title: Stability Indicating Assay Workflow (49 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Vaccine Stability Studies
| Item | Function in Stability Assessment | Example/Notes |
|---|---|---|
| Controlled Stability Chambers | Provides precise, ICH-compliant conditions (temperature, humidity) for real-time and accelerated studies. | Walk-in chambers for 2-8°C; benchtop units for 25°C/60%RH, 40°C/75%RH. |
| Differential Scanning Calorimeter (DSC) | Measures thermal unfolding (Tm) of proteins in vaccine antigens, indicating structural stability. | Critical for adjuvant-antigen complex analysis. |
| Dynamic/Nanoparticle Tracking Light Scattering | Quantifies particle size distribution and detects aggregation/fusion in viral particles or LNPs. | DLS for polydisperse samples; NTA for more precise concentration of sub-populations. |
| Capillary Electrophoresis SDS | High-resolution analysis of protein integrity, quantifying fragments and aggregates in subunit vaccines. | Superior to traditional SDS-PAGE for quantitation and automation. |
| Ion-Pair Reversed-Phase HPLC | Separates and quantifies intact vs. degraded (fragmented) mRNA, the critical quality attribute for mRNA vaccines. | Uses ion-pairing agents like TEA/HEA. |
| Ribogreen Fluorescence Assay | Quantifies total and free mRNA to calculate encapsulation efficiency of LNPs, a marker of particle integrity. | Requires careful lysis protocol standardization. |
| Capture ELISA with Conformational Antibodies | Measures antigenic potency by specifically binding to native, quaternary epitopes that elicit neutralizing antibodies. | Must use a well-characterized, lot-controlled antibody pair. |
| Plaque Assay / TCID50 Reagents | The gold-standard for determining infectious titer of live-attenuated or viral vector vaccines post-stress. | Cell lines, overlay media, staining solutions. |
Application Note: This document provides a comparative analysis of scale-up bottlenecks in traditional egg-based and modern cell-based viral vaccine manufacturing. Framed within the historical progression from Jennerian inoculation to contemporary mRNA platforms, it details the technical constraints, timelines, and protocols defining each method. The focus is on influenza vaccine production as the primary case study, given its utilization of both technologies.
Quantitative Comparison of Manufacturing Bottlenecks
Table 1: Key Bottleneck Parameters in Influenza Vaccine Manufacturing
| Parameter | Egg-Based Manufacturing | Cell-Based (MDCK or Vero Cells) Manufacturing |
|---|---|---|
| Primary Bottleneck | Supply & quality of specific pathogen-free (SPF) eggs; egg-adaptive viral mutations. | Bioreactor capacity; cell bank expansion; serum-free media cost. |
| Typical Lead Time | 6-8 months for seasonal strain update. | 5-7 months for seasonal strain update. |
| Batch Production Time | ~12 weeks (from egg order to purified antigen). | ~10 weeks (from thawing cell bank to purified antigen). |
| Scalability Limit | Linear; requires millions of embryonated eggs. | Exponential; limited by bioreactor volume and cell growth kinetics. |
| Yield Variability | High; depends on virus strain growth in eggs. | More consistent; controlled environment. |
| Regulatory Timeline (New Strain) | Established but includes ~2 months for egg adaptation & seed generation. | Streamlined for pre-approved cell lines; no egg-adaptation needed. |
| Allergen Risk | Present (ovalbumin). | Negligible. |
Table 2: Timeline Breakdown for Seasonal Influenza Vaccine (From WHO Strain Selection to Fill/Finish)
| Process Step | Egg-Based (Duration) | Cell-Based (Duration) |
|---|---|---|
| Virus Seed Generation | 3-4 weeks (requires egg-adaptation) | 2-3 weeks (no adaptation needed) |
| Upstream Production | 8-10 days (inoculation & incubation in eggs) | 7-10 days (bioreactor infection & harvest) |
| Harvest | 2-3 days (manual/allantoic fluid aspiration) | 1-2 days (automated bioreactor harvest) |
| Downstream Purification | 4-5 weeks (ultrafiltration, centrifugation, inactivation) | 3-4 weeks (similar but more consistent feedstock) |
| Total Core Manufacturing | ~12 weeks | ~10 weeks |
Detailed Experimental Protocols
Protocol 2.1: Egg-Based Influenza Virus Seed Adaptation and Amplification
Objective: To generate a high-growth reassortant or egg-adapted virus seed suitable for large-scale propagation in embryonated chicken eggs.
Materials:
- World Health Organization (WHO)-recommended influenza virus strain.
- Specific Pathogen-Free (SPF) embryonated chicken eggs (9-11 days old).
- Class II Biological Safety Cabinet.
- Egg candler, drill, and sealing wax/tape.
- Sterile syringes and 18-25 gauge needles.
- Phosphate-Buffered Saline (PBS) with antibiotics (Pen/Strep).
- Refrigerated centrifuge.
Methodology:
- Virus Inoculation: Candle eggs to identify viable embryo and air sac. Drill a small hole at the top (air sac) and over the allantoic cavity. Using a sterile syringe, inoculate 0.1-0.2 mL of virus stock (diluted in PBS) into the allantoic cavity. Seal hole with wax/tape.
- Incubation: Incubate inoculated eggs at 33-35°C for 48-72 hours with appropriate humidity. Candle daily to monitor embryo viability.
- Harvest: Chill eggs at 4°C for ≥4 hours to embryo and constrict blood vessels. Aseptically open the allantoic cavity and harvest the allantoic fluid using a sterile pipette or syringe.
- Clarification: Centrifuge harvested fluid at 1000-2000 x g for 10 min at 4°C to remove debris. Aliquot and store supernatant (crude virus stock) at -80°C.
- Seed Adaptation: Repeat steps 1-4 for 3-5 serial passages in eggs, monitoring hemagglutination (HA) titer at each passage. Select the passage with the highest HA titer for the master virus seed (MVS).
- MVS Characterization: Titrate MVS (TCID50, EID50), sequence HA gene to confirm egg-adaptive mutations (e.g., T160K, L194P in H3N2), and ensure sterility.
Protocol 2.2: Cell-Based Influenza Virus Production in a Stirred-Tank Bioreactor
Objective: To produce influenza virus antigen using MDCK cells in a serum-free, microcarrier-based bioreactor system.
Materials:
- MDCK.SUS2 or similar suspension-adapted cell line.
- Serum-free, protein-free cell culture medium.
- Bioreactor (e.g., 50L stirred-tank) with DO, pH, temperature controls.
- Microcarriers (e.g., Cytodex 1) if using adherent cells.
- Virus seed (cell-derived, titered).
- Trypsin-TPCK (for HA cleavage of influenza).
- Benzonase endonuclease.
- Depth filtration and tangential flow filtration (TFF) systems.
Methodology:
- Bioreactor Preparation: Clean, sterilize (SIP), and calibrate the bioreactor. Fill with basal medium.
- Cell Expansion: Inoculate bioreactor with cells from a N-1 expansion bioreactor at a target viable cell density (VCD) of ~0.5 x 10^6 cells/mL. Maintain at 37°C, pH 7.2, DO at 40% saturation. Allow cells to grow to a target VCD of 2.5-3.0 x 10^6 cells/mL.
- Virus Infection: Cool bioreactor to 33-35°C. Infect cells at a low multiplicity of infection (MOI) of 0.001-0.01 using cell-derived virus seed. Add TPCK-treated trypsin (1-5 µg/mL) to facilitate HA cleavage.
- Harvest Phase: Maintain culture for 48-96 hours post-infection. Monitor metabolite consumption (glucose, glutamine) and viral HA titer. Harvest when cell viability drops significantly (~50%) or HA titer plateaus.
- Clarification & Nuclease Treatment: Transfer harvest to a hold vessel. Add Benzonase (≥50 U/mL) and incubate 2-6 hours at 25°C to digest host cell DNA. Clarify using depth filtration (0.2/0.5 µm).
- Concentration & Purification: Concentrate clarified harvest 10-20 fold using TFF with a 300 kDa MWCO membrane. Follow with sucrose gradient ultracentrifugation or chromatography (e.g., anion exchange) for purification. Inactivate using beta-propiolactone or formaldehyde.
Visualizations
Title: Egg-Based Vaccine Production Workflow with Bottlenecks
Title: Cell-Based Vaccine Production Workflow with Bottlenecks
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Cell-Based Vaccine Process Development
| Reagent/Material | Function & Rationale |
|---|---|
| Suspension-Adapted Cell Line (e.g., MDCK.SUS2, AGE1.CR.pIX) | Host cell system capable of growing in serum-free suspension culture, eliminating reliance on microcarriers and improving scalability. |
| Animal-Component Free, Protein-Free Cell Culture Medium | Supports cell growth and virus production while reducing regulatory concerns regarding adventitious agents and lot-to-lot variability. |
| TPCK-Treated Trypsin | Serine protease essential for cleaving influenza hemagglutinin (HA) into HA1 and HA2 subunits, enabling multi-cycle viral replication in cell culture. TPCK treatment inactivates contaminating chymotrypsin. |
| Recombinant Benzonase Endonuclease | Digests host cell DNA and RNA in the harvest, reducing viscosity and improving downstream filtration and chromatography efficiency. |
| Anion Exchange Chromatography Resins (e.g., Capto Q) | High-capacity resins for purifying influenza virus via binding of negatively charged sialic acid receptors or viral surface proteins, replacing sucrose gradient ultracentrifugation. |
| Gamma-Irradiated, Single-Use Bioreactors | Pre-sterilized, disposable culture vessels that eliminate cleaning validation, reduce cross-contamination risk, and accelerate campaign changeover. |
Application Notes
The development of viral vector vaccines, particularly those based on human adenoviruses (HAdVs), represents a pivotal chapter in the historical continuum from Jenner's empirical inoculations to the rational design of mRNA platforms. While these vectors offer high transduction efficiency and robust immunogenicity, two primary challenges impede their universal application: pre-existing immunity (PEI) to the viral vector and inherent reactogenicity. PEI, stemming from prior natural infection with common HAdVs (e.g., HAdV-C5), can neutralize the vector, drastically reducing vaccine potency and transgene expression. Concurrently, the innate immune recognition of viral components triggers reactogenicity—transient inflammatory side effects that, while often manageable, require precise profiling for clinical acceptance and dose optimization.
Recent strategies to circumvent PEI include the engineering of rare serotype vectors (e.g., HAdV-D26, HAdV-B35), chimeric vectors, and non-human adenoviruses (e.g., chimpanzee-derived ChAdOx1). Profiling reactogenicity involves quantifying innate cytokines (IL-6, TNF-α, IFN-γ) and employing systems vaccinology to correlate early molecular signatures with clinical adverse events. The following protocols provide methodologies for quantifying PEI and characterizing reactogenicity profiles in preclinical models.
Protocols
Protocol 1: Quantification of Pre-Existing Adenovirus Neutralizing Antibodies (NAbs) Using a Luciferase-Based Reporter Assay
Objective: To measure serum neutralizing antibody titers against a specific adenovirus serotype (e.g., HAdV-C5, HAdV-D26) that can inhibit vector transduction.
Materials:
- Serum samples (heat-inactivated at 56°C for 30 min).
- Target adenovirus vector expressing firefly luciferase (e.g., HAdV5-Luc).
- Permissive cell line (e.g., HEK293 or A549 cells).
- Cell culture medium and 96-well tissue culture plates.
- Luciferase assay reagent and luminometer.
- Positive control (high-titer anti-adenovirus serum).
- Negative control (naïve serum or medium).
Procedure:
- Serum Dilution: Prepare serial 2-fold dilutions of each test serum in culture medium in a 96-well plate (e.g., 1:20 to 1:2560). Include positive and negative controls.
- Virus Incubation: Add a fixed titer of HAdV5-Luc (e.g., 1x10^8 viral particles/mL) to each serum dilution at a 1:1 ratio. Incubate at 37°C for 1 hour.
- Cell Seeding and Infection: Seed A549 cells at 2x10^4 cells/well in a separate 96-well plate. After serum-virus incubation, transfer the mixture onto the cell monolayer.
- Incubation: Incubate cells at 37°C, 5% CO2 for 24-48 hours.
- Luciferase Assay: Lyse cells and add luciferase substrate. Measure luminescence using a luminometer.
- Data Analysis: Calculate the percentage of neutralization for each serum dilution relative to the negative control (virus-only wells). The neutralizing antibody titer (NT50) is defined as the serum dilution that inhibits 50% of luciferase activity. This can be calculated using non-linear regression (four-parameter logistic curve).
Table 1: Representative Neutralizing Antibody (NAb) Titers (NT50) Against Common Adenovirus Serotypes in Global Cohorts
| Serotype | Cohort Description | Geometric Mean Titer (GMT) | Seroprevalence (%) (>NT50 of 20) | Key Reference (Year) |
|---|---|---|---|---|
| HAdV-C5 | US & European Adults | 250 - 500 | 60 - 85% | Barouch et al., 2011 |
| HAdV-D26 | Sub-Saharan Africa | 80 - 200 | 30 - 50% | Mennechet et al., 2019 |
| ChAdOx1 | Global (Pre-COVID) | <20 | 1 - 5% | Folegatti et al., 2020 |
| HAdV-B35 | Southeast Asia | 100 - 400 | 40 - 70% | Tan et al., 2019 |
Protocol 2: In Vivo Profiling of Vector-Induced Reactogenicity in a Murine Model
Objective: To characterize the innate immune reactogenicity profile following intramuscular administration of an adenoviral vector.
Materials:
- C57BL/6 mice (6-8 weeks old).
- Purified adenovirus vector (e.g., ChAdOx1-nCoV-19) and PBS control.
- ELISA kits for murine IL-6, TNF-α, CXCL1.
- Flow cytometry antibodies: CD45, CD11b, Ly6G, Ly6C, CD80, MHC II.
- RNAlater solution and RNA extraction kits.
- Real-time PCR reagents.
Procedure:
- Vector Administration: Administer a dose range of the vector (e.g., 1x10^8 to 1x10^10 vp) intramuscularly to groups of mice (n=5). Administer PBS to the control group.
- Clinical Monitoring: Monitor mice for local (swelling, redness) and systemic (activity, posture) signs for 72 hours.
- Sample Collection: At defined timepoints (e.g., 6, 24, 48h), collect blood via retro-orbital bleed and dissect the injection site muscle.
- Systemic Cytokine Analysis: Analyze serum for IL-6, TNF-α, and CXCL1 levels by ELISA.
- Local Immune Infiltrate Analysis: Digest muscle tissue to single-cell suspension. Stain for innate immune cells (neutrophils: CD11b+ Ly6G+; inflammatory monocytes: CD11b+ Ly6C+; activated dendritic cells: CD11c+ CD80+ MHC II+) and analyze by flow cytometry.
- Gene Expression Profiling: Isolve RNA from muscle tissue. Perform qRT-PCR for inflammatory markers (e.g., Il6, Tnf, Nlrp3, Ifnb1).
Table 2: Representative Reactogenicity Biomarkers Post Adenovirus Vector Immunization
| Biomarker/Readout | Sample Type | Peak Time Post-Injection | Correlation with Clinical AE | Notes |
|---|---|---|---|---|
| Serum IL-6 | Blood | 6 - 12 hours | Strong for fever, myalgia | Key systemic cytokine |
| Muscle Il1b mRNA | Tissue | 12 - 24 hours | Local inflammation | Indicates inflammasome activation |
| Neutrophil Influx | Tissue (Flow) | 24 - 48 hours | Local swelling/pain | Primary early infiltrate |
| IFN-γ ELISpot | PBMCs/Spleen | 24 - 72 hours | Systemic symptoms | Early antigen-independent response |
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Adenovirus Vector Studies
| Item | Function & Application | Example Product/Catalog |
|---|---|---|
| Adenovirus Purification Kit | Purifies viral particles from lysates via column-based methods; essential for high-titer, endotoxin-low preps. | Adeno-X Maxi Purification Kit (Takara Bio) |
| Ready-to-Use Luciferase Reporter Adenovirus | Validated vector for rapid neutralization assays; eliminates need for in-house virus construction. | Adenovirus (Firefly Luciferase) (Vector Biolabs) |
| Adenovirus Neutralizing Antibody Positive Control | High-titer, standardized anti-serum for assay validation and as a positive control in NAb assays. | Anti-Adenovirus 5 Neutralizing Ab (Merck) |
| Multiplex Cytokine Panel (Mouse) | Quantifies multiple reactogenicity-associated cytokines (IL-6, TNF-α, KC/GRO, IFN-γ) from small serum volumes. | Mouse ProcartaPlex Panel (Thermo Fisher) |
| Myeloid Cell Staining Panel for Flow Cytometry | Antibody cocktail for identifying neutrophils, monocytes, and macrophages in infiltrated tissues (muscle). | Mouse Myeloid Phenotyping Panel (BioLegend) |
| In Vivo Transfection Reagent (Optional) | Enhances adenovirus transduction in vivo for models with low receptor expression; can alter biodistribution. | in vivo-jetPEI (Polyplus) |
Visualizations
Diagram 1: PEI Impact on Vaccine Efficacy
Diagram 2: Innate Immune Pathways Driving Reactogenicity
Diagram 3: In Vivo Reactogenicity Profiling Workflow
The mRNA Disruption: A Comparative Analysis of Efficacy, Speed, and Versatility
The journey from Edward Jenner’s empirical use of cowpox virus in 1798 to the rational design of mRNA-LNP vaccines represents a paradigm shift in vaccinology. This application note details the pivotal experiments and protocols that transformed mRNA from a theoretical therapeutic concept into a clinically validated platform, contextualized within the broader history of viral vaccine development.
Key Quantitative Data Summaries
Table 1: Evolution of mRNA Modifications & Stability (Key In Vitro Findings)
| Parameter | Unmodified mRNA (Control) | N1-Methylpseudouridine (m1Ψ) Modified mRNA | Assay Type | Reference (Example) |
|---|---|---|---|---|
| TLR7/8 Activation (IL-6 release) | High (>1000 pg/mL) | Low (<100 pg/mL) | Human PBMC assay | Karikó et al., 2005 |
| Protein Expression In Vitro (RLU) | 1.0 x 10^6 | 5.2 x 10^6 | Luciferase in HeLa cells | Karikó et al., 2008 |
| In Vivo Protein Expression Half-life | ~2-4 hours | ~6-10 hours | Bioluminescence in mice | Andries et al., 2015 |
| Ribonucleotide Incorporation Efficiency | Baseline | ~95% of Uridine replaced | HPLC-MS | Karikó et al., 2008 |
Table 2: LNP Formulation Characteristics for SARS-CoV-2 mRNA Vaccines
| Component & Property | Moderna mRNA-1273 (Spikevax) | Pfizer-BioNTech BNT162b2 (Comirnaty) | Function |
|---|---|---|---|
| Ionizable Lipid | SM-102 | ALC-0315 | Encapsulation, endosomal release |
| Lipid:MRNA N:P Ratio | ~6:1 | ~3:1 | Complexation efficiency |
| Particle Size (Z-Average) | ~80-100 nm | ~70-85 nm | Delivery, biodistribution |
| Polydispersity Index (PDI) | <0.2 | <0.2 | Batch uniformity |
| Encapsulation Efficiency | >95% | >95% | Protection from RNase |
Detailed Experimental Protocols
Protocol 3.1: In Vitro Transcription (IVT) for Modified mRNA Production
Objective: Synthesize cap-modified, m1Ψ-substituted mRNA. Reagents:
- Template: Linearized plasmid DNA with T7 promoter and poly(A) tail sequence.
- NTPs: ATP, GTP, CTP, N1-Methylpseudouridine-5'-Triphosphate (m1ΨTP).
- Enzymes: T7 RNA Polymerase, Pyrophosphatase.
- Cap Analog: CleanCap AG (3' O-Me-m7G(5')ppp(5')G).
- Buffer: Tris-HCl, MgCl2, Spermidine, DTT.
Procedure:
- Reaction Setup: Combine in nuclease-free tube: 1 µg linearized DNA template, 7.5 mM each NTP (including m1ΨTP), 6 mM CleanCap AG, 1X transcription buffer, 0.5 U/µL T7 RNA Polymerase, 0.1 U/µL Pyrophosphatase. Incubate at 37°C for 2-3 hours.
- DNase I Treatment: Add 2 U of DNase I (RNase-free) per µg template DNA. Incubate 15 min at 37°C.
- mRNA Purification: Purify using LiCl precipitation (add 0.1 volume 3M NaOAc, 0.7 volume isopropanol) or silica membrane-based columns. Elute in nuclease-free water.
- QC Analysis: Measure concentration (A260). Check integrity via capillary electrophoresis (Bioanalyzer). Verify capping efficiency by LC-MS or anti-cap immunoblot.
Protocol 3.2: Microfluidic Formulation of mRNA-LNPs
Objective: Reproducibly formulate mRNA-loaded LNPs via rapid mixing. Reagents:
- Lipid Stock in Ethanol: Ionizable lipid (e.g., ALC-0315), DSPC, Cholesterol, PEG-lipid (e.g., ALC-0159).
- Aqueous Buffer: mRNA at 0.1 mg/mL in citrate buffer (pH 4.0).
- Dialysis Buffer: 1X PBS, pH 7.4.
Procedure:
- Lipid Solution Prep: Dissolve lipids in ethanol at molar ratio (e.g., 50:10:38.5:1.5 for ionizable lipid:DSPC:Chol:PEG-lipid). Total lipid concentration ~12.5 mM.
- Aqueous Solution Prep: Dilute purified mRNA in 10 mM citrate buffer, pH 4.0.
- Microfluidic Mixing: Using a staggered herringbone mixer chip (or T-junction), mix the ethanolic lipid solution and aqueous mRNA solution at a 3:1 flow rate ratio (aqueous:ethanol). Total flow rate 12 mL/min. Collect effluent.
- Buffer Exchange & Dialysis: Immediately dilute LNP formulation in 1X PBS (pH 7.4). Dialyze against >1000 volumes of PBS for 18 hours at 4°C to remove ethanol and establish neutral pH.
- Filtration & Storage: Sterile-filter (0.22 µm). Aliquot and store at -80°C. QC: Measure particle size (DLS), PDI, encapsulation efficiency (RiboGreen assay), and endotoxin levels.
Protocol 3.3: In Vivo Potency & Immunogenicity Assay
Objective: Evaluate humoral and cellular immune response to mRNA-LNP vaccine in mice. Procedure:
- Immunization: Administer 2 µg (dose in 50 µL) of formulated mRNA-LNP intramuscularly to C57BL/6 mice (n=5/group). Prime at Day 0, boost at Day 21.
- Serum Collection: Collect blood via retro-orbital bleed at Days 0, 14, 28, and 42. Isolate serum.
- Humoral Response (ELISA):
- Coat high-binding plates with recombinant target antigen (e.g., SARS-CoV-2 Spike S1 subunit).
- Add serial dilutions of mouse serum. Detect bound IgG using HRP-conjugated anti-mouse IgG. Calculate endpoint titers.
- Cellular Response (ELISpot):
- At Day 28, isolate splenocytes. Plate cells on IFN-γ pre-coated ELISpot plates.
- Stimulate with a peptide pool spanning the target antigen. Develop spots and count using an automated reader.
- Neutralization Assay (Pseudovirus): Incubate serum with lentiviral particles pseudotyped with target viral glycoprotein. Measure reduction in luminescence in susceptible cells (e.g., HEK293T-ACE2) relative to control serum.
Diagrams
Title: mRNA-LNP Workflow: Synthesis to Immune Response
Title: How m1Ψ mRNA Modification Enhances Protein Yield
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for mRNA-LNP Research
| Reagent / Solution | Function / Role in Protocol | Example Vendor / Cat. No. (for reference) |
|---|---|---|
| N1-Methylpseudouridine-5'-Triphosphate (m1ΨTP) | Modified nucleoside replacing UTP during IVT to suppress innate immune recognition and enhance translation. | TriLink BioTechnologies, N-1081 |
| CleanCap Cap Analog (e.g., CleanCap AG) | Co-transcriptional capping agent producing Cap 1 structure (m7GpppNm), essential for high translation efficiency and reduced immunogenicity. | TriLink BioTechnologies, N-7113 |
| Ionizable Cationic Lipid (e.g., DLin-MC3-DMA, SM-102) | Critical LNP component for mRNA encapsulation, endosomal escape via protonation at low pH. | Avanti Polar Lipids, MedChemExpress |
| PEGylated Lipid (e.g., DMG-PEG2000, ALC-0159) | LNP surface stabilizer controlling particle size and pharmacokinetics; shields from non-specific interactions. | Avanti Polar Lipids, 880151 |
| T7 RNA Polymerase, High Yield | Bacteriophage-derived RNA polymerase for efficient, DNA-templated in vitro transcription. | New England Biolabs, M0251 |
| RiboGreen RNA Quantitation Assay | Fluorescent dye-based assay to quantify total vs. free RNA, determining LNP encapsulation efficiency. | Invitrogen, R11490 |
| Microfluidic Device (NanoAssemblr) | Instrument for reproducible, scalable LNP formation via rapid mixing of lipid and aqueous phases. | Precision NanoSystems |
| Luciferase-Encoding Control mRNA (modified) | Positive control for transfection and delivery experiments; enables quantitative protein expression readout. | Aldevron, L-7202 |
| Human TLR7/8 Reporter Cell Line (HEK293) | Cell-based assay system to quantify innate immune activation potential of mRNA formulations. | InvivoGen, hkb-htlr7) |
| Splenocyte Isolation Kit | For harvesting mouse splenocytes to analyze antigen-specific T-cell responses via ELISpot/Flow Cytometry. | STEMCELL Technologies, 19854 |
Application Notes: A Paradigm Shift in Vaccine Development Timeline
The development of SARS-CoV-2 vaccines, specifically mRNA and adenoviral vector platforms, represents a radical departure from historical norms. While the journey from Edward Jenner's smallpox inoculation (1796) to a licensed vaccine typically spanned decades, the COVID-19 pandemic compressed this to under a year. This was not a singular breakthrough but the convergence of decades of foundational research on coronaviruses (SARS-1, MERS), mRNA stabilization (Karikó and Weissman, 2005), and scalable lipid nanoparticle (LNP) delivery systems.
The critical application notes from this effort are:
- Platform Agility: The mRNA platform is sequence-agnostic. Once the viral genome (published Jan 10, 2020) identified the spike (S) protein as the target, candidate vaccine sequences could be designed in silico within days.
- Parallel Processing: Pre-clinical, Phase I/II/III clinical trials, and manufacturing scale-up were conducted in parallel, not sequentially, based on real-time data sharing and regulatory feedback (e.g., FDA's Emergency Use Authorization pathway).
- Real-World Efficacy Correlates: Post-authorization surveillance established real-world effectiveness (RWE) data at unprecedented scale and speed, validating trial results and informing recommendations on boosters and variants.
Table 1: Historical vs. COVID-19 Vaccine Development Timeline Comparison
| Vaccine / Platform | Pathogen | Year of First Use/Concept | Year of Licensure/Authorization | Approx. Development Time | Key Reason for Pace |
|---|---|---|---|---|---|
| Smallpox (Live-virus) | Variola virus | 1796 (Jenner) | ~1950s (Standardization) | >150 years | Empirical observation, no regulatory framework |
| Polio (Inactivated) | Poliovirus | 1955 (Salk) | 1955 | ~45 years (from isolation) | Cell culture technology, large-scale clinical trials |
| HPV (Recombinant VLP) | Human Papillomavirus | 2006 (Gardasil) | 2006 | ~15 years (from VLP proof) | Recombinant protein expression, purification |
| SARS-CoV-2 (mRNA-LNP) | SARS-CoV-2 | 2020 (BNT162b2/mRNA-1273) | 2020 | < 1 year | Prefabricated platform, pandemic urgency, prior research investment |
| SARS-CoV-2 (AdV Vector) | SARS-CoV-2 | 2020 (AZD1222/Ad26.COV2.S) | 2020/2021 | < 1 year | Veteran vector platform (e.g., Ebola), rapid antigen cloning |
Table 2: Key Efficacy/Effectiveness Data from Initial COVID-19 Vaccine Trials
| Vaccine (Platform) | Trial Phase | Efficacy Against Symptomatic COVID-19 (95% CI) | Efficacy Against Severe Disease/Hospitalization | Study Population | Primary Endpoint |
|---|---|---|---|---|---|
| BNT162b2 (mRNA) | Phase III | 95.0% (90.3-97.6) | ~100% (preventive) | >43,000 participants | Lab-confirmed COVID-19 at ≥7 days post-dose 2 |
| mRNA-1273 (mRNA) | Phase III | 94.1% (89.3-96.8) | 100% (preventive) | >30,000 participants | Lab-confirmed COVID-19 at ≥14 days post-dose 2 |
| AZD1222 (ChAdOx1) | Phase III | 74.0% (65.3-80.5) | 100% (preventive) | ~32,000 participants | Lab-confirmed symptomatic COVID-19 |
| Ad26.COV2.S (Ad26) | Phase III | 66.9% (59.0-73.4) | 76.7% (54.6-89.1) against severe/critical disease | ~40,000 participants | Lab-confirmed moderate to severe/critical COVID-19 at ≥14/28 days post-dose |
Detailed Protocols
Protocol 2.1:In VitroTranscript (IVT) Synthesis and LNP Formulation of mRNA Vaccine (Simplified Research Scale)
Aim: To produce research-grade mRNA-LNP encoding the SARS-CoV-2 Spike protein.
I. Template DNA Preparation (Circular Plasmid)
- Clone a gene cassette into a plasmid vector (e.g., pVAX1) containing: T7 promoter, 5' UTR, Kozak sequence, SARS-CoV-2 S-2P gene (mutations for prefusion stabilization), 3' UTR, and poly(A) tail (~120 nucleotides).
- Amplify the plasmid in E. coli (e.g., DH5α) and purify using an endotoxin-free maxiprep kit.
- Linearize the plasmid downstream of the poly(A) tail using a restriction enzyme (e.g., PmeI). Verify complete linearization by agarose gel electrophoresis.
- Purify the linearized DNA template using a PCR cleanup kit. Determine concentration (ng/μL) and purity (A260/A280 ~1.8-2.0) via spectrophotometry.
II. In Vitro Transcription (IVT) and Capping
- Assemble IVT reaction (100 μL scale):
- Linearized DNA template: 1 μg
- NTPs (including 5mCTP and pseudo-UTP): 7.5 mM each
- T7 RNA Polymerase Buffer (10X): 10 μL
- T7 RNA Polymerase: 0.5 μL
- Pyrophosphatase (optional): 0.1 μL
- RNase Inhibitor: 1 μL
- Nuclease-free water to 100 μL
- Incubate at 37°C for 2-4 hours.
- Add DNase I (1 μL, RNase-free) and incubate at 37°C for 15 min to digest the DNA template.
- Perform Co-transcriptional Capping using CleanCap Reagent (Trilink) as per manufacturer's instructions for >95% capping efficiency.
- Purify mRNA using magnetic bead-based purification (e.g., RNA Clean & Concentrator kits). Elute in nuclease-free water.
- Quality Control: Analyze integrity via capillary electrophoresis (e.g., Fragment Analyzer), determine concentration, and check for dsRNA contamination.
III. Lipid Nanoparticle (LNP) Formulation (Microfluidic Mixing)
- Prepare Lipid Mixture in Ethanol:
- Ionizable cationic lipid (e.g., ALC-0315, SM-102): 50 mol%
- Phospholipid (e.g., DSPC): 10 mol%
- Cholesterol: 38.5 mol%
- PEGylated lipid (e.g., ALC-0159, DMG-PEG 2000): 1.5 mol%
- Dissolve in ethanol to a total lipid concentration of ~12.5 mM.
- Prepare Aqueous Phase: Dilute purified mRNA in citrate buffer (pH 4.0) to 0.1 mg/mL.
- Formulate using a microfluidic mixer (e.g., NanoAssemblr Ignite):
- Set flow rate ratio (aqueous:ethanol) to 3:1.
- Set total flow rate to achieve a mixing time of <10 ms.
- Collect formulated LNPs in a vessel.
- Dialyze against PBS (pH 7.4) for 4 hours at 4°C to remove ethanol and establish neutral pH.
- Filter through a 0.22 μm sterile filter.
- Quality Control: Measure particle size and PDI via DLS (~80-100 nm), mRNA encapsulation efficiency (using dye exclusion assay), and concentration.
Protocol 2.2: Pseudovirus Neutralization Assay for Vaccine Sera Evaluation
Aim: To quantify neutralizing antibody titers in serum from vaccinated individuals against SARS-CoV-2 Spike protein.
Materials:
- HEK293T/17 cells (ATCC CRL-11268)
- Lentiviral packaging plasmids (psPAX2, pMD2.G)
- Reporter plasmid (e.g., pLenti-CMV-Luciferase-Puro)
- Spike-expressing plasmid (e.g., pcDNA3.1-SARS2-SΔ19)
- Serum samples (heat-inactivated at 56°C for 30 min)
- Dulbecco's Modified Eagle Medium (DMEM), Fetal Bovine Serum (FBS), Polybrene (8 μg/mL)
- Luciferase assay reagent (e.g., Bright-Glo)
- 96-well tissue culture plates (white, clear bottom)
Procedure:
- Pseudovirus Production (Day 1): Co-transfect HEK293T cells in a 10cm dish with packaging plasmids (psPAX2), reporter plasmid, and Spike plasmid using a transfection reagent (e.g., PEI). Replace medium after 6-8 hours.
- Harvest Virus (Day 2 & 3): Collect supernatant at 48 and 72 hours post-transfection, filter through 0.45μm, aliquot, and store at -80°C. Titrate on HEK293T-ACE2 cells.
- Neutralization Assay (Day 4):
- Serum Dilution: Perform 3-fold serial dilutions of heat-inactivated serum in culture medium in a 96-well plate.
- Virus Incubation: Mix equal volumes of diluted serum and pseudovirus (pre-titered to give ~1e6 RLU) and incubate at 37°C for 1 hour.
- Cell Infection: Seed HEK293T-ACE2 cells at 10,000 cells/well in a separate plate. Add the serum-virus mixture to cells in the presence of Polybrene.
- Incubate: Incubate for 48-72 hours at 37°C, 5% CO2.
- Readout (Day 6/7): Aspirate medium, add luciferase reagent, and measure luminescence.
- Analysis: Calculate % neutralization as:
[1 - (RLU with serum / RLU without serum)] * 100. Determine NT50 (serum dilution that gives 50% neutralization) using a 4-parameter logistic curve fit.
Visualization Diagrams
Title: mRNA Vaccine Development Workflow (2020)
Title: mRNA-LNP Vaccine Immunological Mechanism
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for mRNA Vaccine R&D
| Reagent / Material | Function / Role | Example Product / Component |
|---|---|---|
| N1-Methylpseudouridine (m1Ψ) | Modified nucleoside; replaces uridine to decrease innate immune sensing (TLR7/8) and increase translational efficiency. | Trilink BioTechnologies CleanCap m1Ψ |
| Ionizable Cationic Lipid | Critical LNP component; positively charged at low pH (aids encapsulation), neutral in blood (reduces toxicity), facilitates endosomal escape. | SM-102 (Moderna), ALC-0315 (BioNTech/Pfizer) |
| PEGylated Lipid | LNP component; stabilizes particle, modulates size, reduces aggregation, and influences pharmacokinetics. Can induce anti-PEG antibodies. | DMG-PEG 2000, ALC-0159 |
| T7 RNA Polymerase | High-yield bacteriophage-derived polymerase for in vitro transcription of mRNA from DNA template. | HiScribe T7 ARCA mRNA Kit (NEB) |
| Cap Analog | Ensures proper 5' capping of mRNA, critical for ribosome recognition, stability, and reducing immune activation. | CleanCap AG (Trilink) for co-transcriptional capping. |
| RNase Inhibitor | Protects mRNA during synthesis and handling from degradation by ribonucleases. | Recombinant RNase Inhibitor (Takara) |
| HEK293T Cells | Workhorse cell line for pseudovirus production due to high transfection efficiency and robust growth. | ATCC CRL-3216 |
| Human ACE2-expressing Cell Line | Essential for SARS-CoV-2 pseudovirus or live virus neutralization assays. | HEK293T-hACE2 (BEI Resources) |
| Luciferase Reporter Plasmid | Quantifiable reporter gene (e.g., Fluc) for packaging into pseudoviruses to measure infection/neutralization. | pLenti-CMV-Luc-Puro (Addgene) |
| Microfluidic Mixer | Enables reproducible, scalable mixing for LNP formulation via rapid turbulent mixing of lipid and aqueous phases. | NanoAssemblr (Precision NanoSystems) |
Application Notes: Historical Context & Rationale The evolution of viral vaccines, from Edward Jenner’s empirical use of cowpox virus (vaccinia) to confer cross-protection against smallpox, to the modern era of genetic platforms, represents a journey of increasing molecular precision. Live-attenuated and inactivated pathogens gave way to subunit vaccines, followed by viral vectors—a technology leveraging engineered viruses (e.g., adenovirus, vaccinia) as delivery shuttles for antigen genes. The COVID-19 pandemic catalyzed the first widespread deployment of two novel platforms: mRNA and adenovirus-vectored (AdV) vaccines. This application note provides a framework for the direct comparative assessment of their immunogenicity and efficacy, anchoring this analysis within the historical continuum of vaccine development. The protocols herein are designed for researchers conducting head-to-head evaluations in preclinical and clinical study contexts.
Table 1: Comparative Platform Characteristics
| Feature | mRNA Platform (e.g., BNT162b2, mRNA-1273) | Adenovirus-Vector Platform (e.g., ChAdOx1, Ad26.COV2.S) |
|---|---|---|
| Key Components | Nucleoside-modified mRNA, lipid nanoparticles (LNPs) | Non-replicating adenovirus vector (chimpanzee/human), transgene cassette |
| Antigen Production | Host cell cytoplasm translation of encoded Spike protein | Host cell nucleus transcription/translation of encoded Spike protein |
| Primary Immune Stimuli | Translated antigen + LNP adjuvant effect + mRNA innate sensing | Translated antigen + adenovirus vector innate immune sensing |
| Dosing Regimen | Two doses, 3-4 weeks apart | Single dose (J&J) or two doses (AstraZeneca, 4-12 weeks apart) |
| Storage | Ultra-cold chain (-80°C to -60°C) | Refrigeration (2°C to 8°C) |
| Key Historical Precedent | Decades of incremental nucleic acid vaccine research | Viral vector development (e.g., Ebola vaccine, Ervebo) |
Table 2: Summary of Reported Efficacy & Immunogenicity (Selected Clinical Trial & Real-World Data)
| Parameter | mRNA Vaccines (2-dose series) | Adenovirus-Vectored Vaccines (Primary series) |
|---|---|---|
| Efficacy vs. Symptomatic COVID-19 (Initial Trials) | 94-95% (Pfizer, Moderna) | 66-79% (J&J), ~76% (AstraZeneca) |
| Efficacy vs. Severe Disease/Hospitalization | >95% | ~85-100% |
| Peak Neutralizing Antibody Titers | High (Reference range) | Moderate (Approx. 2-5x lower than mRNA) |
| CD4+ T-cell Response | Strong Th1-biased | Strong Th1-biased |
| CD8+ T-cell Response | Moderate | Robust (Advantage of viral vector) |
| Onset of Protection | ~12-14 days post-dose 1 | ~14-28 days post-single dose |
Experimental Protocols
Protocol 1: Head-to-Head Humoral Immunogenicity Assay (ELISA & Pseudovirus Neutralization) Objective: Quantify and compare antigen-binding and functional neutralizing antibody titers induced by different vaccine platforms in serum samples. Materials: See "The Scientist's Toolkit" below. Method:
- Serum Collection: Collect sera from vaccinated subjects at pre-defined timepoints (e.g., pre-vaccination, day 28 post-priming, post-boost).
- Anti-Spike IgG ELISA: a. Coat high-binding 96-well plates with recombinant SARS-CoV-2 Spike protein (2 µg/mL in PBS, 100 µL/well) overnight at 4°C. b. Block with 5% non-fat milk in PBS-T for 2 hours at room temperature (RT). c. Add serially diluted serum samples (3-fold dilutions in blocking buffer) and incubate for 2 hours at RT. Include a standard curve (WHO International Standard for anti-SARS-CoV-2 immunoglobulin). d. Wash and add HRP-conjugated anti-human IgG detection antibody. Incubate 1 hour at RT. e. Develop with TMB substrate, stop with 1M H2SO4, and read absorbance at 450 nm. f. Calculate endpoint titers or binding antibody units (BAU)/mL relative to the standard.
- Pseudovirus Neutralization Assay (pVNT): a. Generate SARS-CoV-2 Spike-pseudotyped lentiviral particles expressing a reporter gene (e.g., luciferase). b. Incubate serial serum dilutions (1:20 starting, 3-fold) with a fixed dose of pseudovirus (200 TCID50) for 1 hour at 37°C. c. Add mixture to HEK293T-ACE2 cells in a 96-well plate. Incubate for 48-72 hours. d. Lysc cells and measure reporter activity. The neutralization titer (ID50 or ID80) is the serum dilution that inhibits 50% or 80% of reporter signal compared to virus-only controls.
Protocol 2: Cellular Immunogenicity by IFN-γ ELISpot Objective: Quantify vaccine-induced Spike-specific T-cell responses. Method:
- PBMC Isolation: Isolate peripheral blood mononuclear cells (PBMCs) from heparinized blood via density gradient centrifugation.
- Plate Coating: Coat 96-well PVDF membrane plates with anti-human IFN-γ capture antibody (15 µg/mL in PBS) overnight at 4°C.
- Blocking & Seeding: Block plate with RPMI-10% FBS for 2 hours at RT. Seed PBMCs (2.5 x 10^5 cells/well) in duplicate/triplicate.
- Stimulation: Stimulate cells for 24-48 hours at 37°C with:
- Negative control: Media alone.
- Positive control: PHA (5 µg/mL).
- Test: Overlapping peptide pools spanning the SARS-CoV-2 Spike protein (2 µg/mL per peptide).
- Detection: Wash, add biotinylated anti-IFN-γ detection antibody, followed by streptavidin-ALP. Develop with BCIP/NBT substrate.
- Analysis: Count spot-forming units (SFU) using an automated ELISpot reader. Results expressed as SFU per million PBMCs (background subtracted).
Visualizations
Diagram Title: Antigen Presentation Pathways of mRNA vs. AdV Vaccines
Diagram Title: Comparative Immunogenicity Study Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function in Protocols | Example/Note |
|---|---|---|
| Recombinant SARS-CoV-2 Spike Protein (Trimeric) | Coating antigen for ELISA to quantify binding antibodies. | Essential for standardizing serology; available from multiple suppliers (e.g., Acro Biosystems, Sino Biological). |
| WHO International Standard for anti-SARS-CoV-2 Ig | Reference serum for calibrating ELISA results to Binding Antibody Units (BAU/mL). | Enables cross-laboratory comparability (NIBSC code: 20/136). |
| SARS-CoV-2 Pseudovirus Neutralization Kit | Contains pre-made pseudovirus and susceptible cells for safe, BSL-2 assessment of neutralization. | Commercial kits streamline Protocol 1 (e.g., from InvivoGen, Integral Molecular). |
| Overlapping Spike Peptide Pools (15-mers) | Stimulate Spike-specific T-cells in ELISpot or intracellular cytokine staining (ICS) assays. | Cover entire Spike protein; often separated into subpools (S1, S2). |
| Human IFN-γ ELISpot Kit | Pre-coated plates and optimized reagents for quantifying T-cell responses (Protocol 2). | Available from Mabtech, BD Biosciences, R&D Systems. |
| ACE2-Expressing Cell Line | Critical for pseudovirus neutralization and live virus neutralization assays. | HEK293T-ACE2 or equivalent. |
| Ultra-Low Temperature Freezer (-80°C) | For long-term storage of biological samples (serum, PBMCs) and mRNA vaccine reference materials. | Preserves sample integrity for longitudinal studies. |
The trajectory of vaccine development, from Jenner's empirical use of cowpox to the rational design of mRNA platforms, represents a paradigm shift towards programmable biologics. This application note details protocols for assessing the core advantage of mRNA vaccines: their rapid adaptability against viral evolution and emergent threats, contextualized within this historical progression.
Application Note: Rapid Antigen Switch & Immunogenicity Profiling
Objective: To quantitatively compare the immunogenic performance of an updated mRNA-LNP construct encoding a variant antigen against its predecessor.
Key Quantitative Data Summary:
Table 1: In Vivo Immunogenicity Metrics (Murine Model, Day 28 Post-Prime)
| Metric | Original Construct (WA1 Spike) | Updated Construct (Omicron BA.5 Spike) | Assay |
|---|---|---|---|
| GMT of Anti-Spike IgG (EU/mL) | 1.2 x 10⁵ | 1.5 x 10⁵ | ELISA |
| Neutralizing Titer (ID₅₀) vs. Homologous Virus | 850 | 1100 | PRNT |
| Neutralizing Titer (ID₅₀) vs. Heterologous Virus (BA.5) | 120 | 950 | PRNT |
| CD8+ T-cell Response (SFU/10⁶ splenocytes) | 320 | 310 | ELISpot |
| Rate of Local Reactogenicity (Grade 1-2) | 15% | 18% | Clinical Scoring |
Table 2: In Vitro Expression & Characterization Data
| Parameter | Original Construct | Updated Construct | Method |
|---|---|---|---|
| In Vitro Translation Yield (μg/mL) | 45.2 ± 3.1 | 43.7 ± 4.5 | Cell-free system |
| Correct Folding Confirmation | 92% | 90% | Flow Cytometry (Conformational Ab) |
| LNP Particle Size (nm) | 84.3 ± 0.5 | 85.1 ± 0.6 | DLS |
| mRNA Purity (% full-length) | 98.5% | 98.3% | cIEF |
Protocol 1: Rapid Template Switching for Prototype mRNA Synthesis
Method:
- Bioinformatic Antigen Design: Input variant spike protein sequence (e.g., from GISAID). Identify key mutations (e.g., RBD, NTD). Use structural modeling software (e.g., AlphaFold2) to confirm preserved folding.
- DNA Template Generation: a. For plasmid-based: Clone optimized gene into validated pUC19-based IVT vector backbone containing T7 promoter and 120-nt poly(A) tail via Gibson Assembly. Transform into DH5α E. coli. b. For PCR-based: Design forward primer with T7 promoter sequence (5'-TAATACGACTCACTATAGGG-3') followed by gene-specific 18-25 nt. Use high-fidelity polymerase (e.g., Q5) to amplify template from synthetic dsDNA fragment.
- mRNA Synthesis via IVT: a. Prepare 100 μL IVT reaction: 1 μg linearized DNA template, 10 μL NTP mix (25mM each), 10 μL 10X T7 Reaction Buffer, 2 μL T7 RNA Polymerase, 1.5 μL Pyrophosphatase, 2 μL RNaseOUT. Incubate at 37°C for 2 hours. b. Add 2 μL DNase I (RNase-free), incubate 15 min at 37°C.
- mRNA Capping & Purification: a. Perform co-transcriptional capping using CleanCap Reagent AG (3' OMe). b. Purify mRNA via silica-membrane spin columns. Elute in nuclease-free water. c. Quantify by Nanodrop (A260/A280 ~2.0). Assess integrity via Fragment Analyzer (capillary electrophoresis).
Protocol 2: Parallelized LNP Formulation & In Vitro Screening
Method:
- Microfluidic LNP Formulation: a. Prepare Ethanol Phase: mRNA in 10 mM citrate buffer (pH 3.0) mixed with ionizable lipid (e.g., SM-102), DSPC, cholesterol, and PEG-lipid at molar ratio 50:10:38.5:1.5. b. Prepare Aqueous Phase: 1X PBS (pH 7.4). c. Use a staggered herringbone micromixer. Flow rates: Aqueous Phase at 12 mL/min, Ethanol Phase at 4 mL/min. Collect effluent into a vessel containing 50 mL of 1X PBS (pH 7.4).
- Tangential Flow Filtration (TFF): Concentrate and dialyze LNP solution against 1X PBS using a 100 kDa MWCO cassette. Sterile filter (0.22 μm).
- In Vitro Transfection & Expression Analysis: a. Seed HEK-293T cells in 96-well plate at 2x10⁴ cells/well. b. Transfect with LNP doses equivalent to 10-100 ng mRNA/well. c. At 24h post-transfection, assay for: Expression (luciferase reporter or flow cytometry for surface antigen), Cell Viability (MTS assay), and Innate Immune Activation (qPCR for IFN-β, ISG15).
Protocol 3: Comparative In Vivo Immunogenicity Assessment
Method:
- Animal Immunization: Group BALB/c mice (n=8-10 per group). Administer 2 μg mRNA-LNP (in 50 μL PBS) via intramuscular injection into the tibialis anterior muscle on Day 0 and Day 21.
- Serum Collection & Antibody Titer Analysis: Collect serum on Days 0, 14, 28, and 42. a. Perform ELISA: Coat high-binding plates with 2 μg/mL recombinant spike protein (original & variant). Use serial serum dilutions. Detect with anti-mouse IgG-HRP. Calculate geometric mean titer (GMT). b. Perform Pseudovirus Neutralization Test (PRNT): Incubate serum dilutions with VSV-ΔG-luciferase pseudotyped with variant spike. Infect HEK-293T-ACE2 cells. Measure luciferase signal at 48h. Calculate 50% inhibitory dilution (ID₅₀).
- Cell-Mediated Immunity Assay (ELISpot): Harvest splenocytes at Day 28. Stimulate 2x10⁵ cells/well with peptide pools (15-mers overlapping by 11) spanning the spike protein. Perform murine IFN-γ ELISpot per manufacturer protocol. Count spot-forming units (SFU).
Visualizations
Title: mRNA Vaccine Platform Rapid Prototyping Workflow
Title: mRNA Vaccine Induced Adaptive Immune Signaling Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for mRNA Vaccine Prototyping
| Reagent / Material | Function / Purpose | Example Vendor/Product |
|---|---|---|
| Ionizable Cationic Lipid | Critical LNP component for mRNA encapsulation and endosomal escape. Enables high transfection efficiency. | SM-102, ALC-0315 (from commercial LNP kits) |
| CleanCap Reagent | Co-transcriptional capping analog for producing Cap 1 structure mRNA, enhancing translation and reducing immunogenicity. | TriLink BioTechnologies |
| T7 RNA Polymerase | High-yield phage polymerase for in vitro transcription (IVT) of mRNA from DNA template. | New England Biolabs (HiScribe T7) |
| N1-Methylpseudouridine (m1Ψ) | Modified nucleoside triphosphate for IVT. Replaces uridine to decrease innate immune sensing and increase protein yield. | Thermo Fisher Scientific |
| Microfluidic Mixer Device | For reproducible, scalable formation of mRNA-LNPs via rapid mixing of aqueous and ethanol phases. | Precision Nanosystems (NanoAssemblr) |
| Pseudovirus Neutralization Assay Kit | Pre-packaged system for generating spike-pseudotyped viruses and measuring neutralizing antibodies. | Integral Molecular (Pseudotype Systems) |
| Murine IFN-γ ELISpot Kit | Pre-coated plates and reagents for quantifying antigen-specific T-cell responses from murine splenocytes. | Mabtech |
| Codon Optimization Software | Cloud-based algorithm for optimizing mRNA sequence for human codon usage and structural features. | IDT Codon Optimization Tool, Genscript OptimumGene |
1. Introduction: A Historical Context The trajectory from Jenner’s cowpox inoculations to contemporary mRNA vaccines represents a paradigm shift in antigen design and delivery. However, each technological leap has confronted the enduring triad of challenges: achieving durable protective immunity, maintaining product stability across supply chains, and ensuring manufacturing affordability at scale. This application note details current experimental approaches to these frontiers, providing actionable protocols for researchers.
2. Durability: Beyond Peak Neutralizing Antibody Titers Long-term protection requires the generation of high-quality, long-lived plasma cells (LLPCs) and memory B/T cells. A primary limitation is the rapid waning of humoral immunity observed with some modern platforms, notably against respiratory pathogens.
Table 1: Comparative Durability Metrics from Recent Clinical & Preclinical Studies
| Vaccine Platform (Target) | Median Peak nAb Titer (IU/mL/IC50) | Fold-Decrease at 6-8 Months | Key Correlate of Durability |
|---|---|---|---|
| mRNA-LNP (SARS-CoV-2 WT) | 1000-1400 | 8-10x | Tfh cell frequency & GC persistence |
| Protein/Adjuvant (SARS-CoV-2) | 500-800 | 4-6x | Bone marrow LLPC abundance |
| Adenoviral Vector (SARS-CoV-2) | 400-600 | 10-12x | Memory B cell somatic hypermutation |
| Live-Attenuated (Influenza) | N/A | N/A | Tissue-resident memory T cells (Trm) |
Experimental Protocol 2.1: Longitudinal Analysis of Germinal Center (GC) Reactions Objective: Quantify the magnitude and duration of GC B cell and T follicular helper (Tfh) cell responses following immunization. Materials:
- Animal Model: C57BL/6 mice, 6-8 weeks old.
- Immunogen: Test vaccine formulated per study design.
- Adjuvant: SMNP (Saponin/MPLA Nanoparticle) or poly(I:C) for protein antigens.
- Reagents: Fluorescently labeled anti-CD4, anti-CD19, anti-GL7, anti-CD95, anti-PD-1, anti-CXCR5 antibodies; FACS buffer. Method:
- Immunization: Prime animals (n=8/group) intramuscularly (i.m.) on Day 0. Boost on Day 21.
- Tissue Harvest: Euthanize cohorts at Days 7, 14, 28, 56, and 120 post-boost.
- Cell Isolation: Dissect draining lymph nodes (dLNs) and spleens. Prepare single-cell suspensions using 70µm cell strainers.
- Flow Cytometry Staining: Stain cells for surface markers (30 min, 4°C). Fix and permeabilize for intracellular staining if required.
- Analysis: Acquire data on a high-parameter cytometer. Gate on live CD19+ GL7+ CD95+ for GC B cells and live CD4+ PD-1hi CXCR5+ for Tfh cells.
- Data Presentation: Report GC B/Tfh cell frequency as % of parent lymphocyte population over time. Perform statistical analysis (e.g., two-way ANOVA).
Diagram 1: Pathway to Durable Humoral Immunity
3. Thermostability: Overcoming the Cold Chain The requirement for ultra-low temperature storage (-80°C to -20°C) for mRNA-LNP and some viral vector vaccines imposes significant logistical and economic burdens in low-resource settings.
Experimental Protocol 3.1: Formulation Screening for Lyophilized mRNA-LNP Objective: Identify optimal cryo/lyo-protectant formulations to preserve mRNA integrity and LNP structure upon lyophilization. Materials:
- Test Formulation: mRNA-LNP (e.g., encoding luciferase).
- Excipients: Sucrose, Trehalose, Mannitol, Polyethylene glycol (PEG), Tromethamine buffer.
- Equipment: Freeze dryer, Dynamic Light Scattering (DLS) instrument, Nanoparticle Tracking Analyzer (NTA), RT-PCR machine. Method:
- Formulation: Mix mRNA-LNP with excipient combinations in 1:1 to 1:5 (w/w) ratios in glass vials (n=4/formulation).
- Lyophilization: Flash-freeze in liquid nitrogen. Perform primary drying at -40°C for 48h under vacuum (<0.1 mBar). Conduct secondary drying at 25°C for 24h.
- Reconstitution: Add nuclease-free water, gently vortex, and incubate for 5 min at RT.
- Characterization:
- Physical: DLS/NTA for particle size (nm), PDI, and concentration.
- Chemical: Ribogreen assay for mRNA encapsulation efficiency (%).
- Functional: In vitro transfection of HEK293T cells; measure luminescence (RLU) at 24h post-transfection.
- Stability Testing: Store lyophilized cakes at 4°C, 25°C, and 37°C for 1, 3, and 6 months. Repeat characterization at each timepoint.
Table 2: Exemplar Data for Lyophilized mRNA-LNP Stability (3 Months)
| Excipient Formulation | Storage Temp. | Size (nm) Post-Reconstitution | PDI | Encapsulation (%) | Relative Potency (%) |
|---|---|---|---|---|---|
| Sucrose:Tromethamine (4:1) | 4°C | 82 ± 3 | 0.08 | 95.2 ± 1.1 | 98 ± 5 |
| Sucrose:Tromethamine (4:1) | 25°C | 85 ± 5 | 0.09 | 94.1 ± 2.0 | 95 ± 7 |
| Trehalose:PEG (3:2) | 25°C | 120 ± 15 | 0.25 | 88.5 ± 3.5 | 80 ± 12 |
| Unformulated (Liquid, -80°C) | -80°C | 80 ± 2 | 0.06 | 96.5 ± 0.5 | 100 (Ref) |
4. Cost of Goods (COGs): Innovations for Affordable Scale-Up COGs are driven by raw material costs (e.g., proprietary lipids, enzymes), process complexity, and fill-finish steps. Reducing COGs is critical for global equity.
Experimental Protocol 4.1: In Vitro Transcription (IVT) Reaction Optimization for mRNA Yield & Capping Efficiency Objective: Maximize yield and 5' Cap-1 structure formation while reducing consumable costs per mg of mRNA. Materials:
- DNA Template: Linearized plasmid or PCR product with T7 promoter.
- NTPs: CleanCap AG analog (Trilink) or enzymatic capping system.
- Enzymes: T7 RNA polymerase, recombinant RNase inhibitor, inorganic pyrophosphatase.
- Purification: Tangential Flow Filtration (TFF) system vs. silica membrane columns. Method:
- IVT Setup: Set up 10 mL reactions varying: NTP concentration (4-8 mM), Mg2+ concentration (20-40 mM), T7 polymerase concentration (0.05-0.2 mg/mL). Incubate at 37°C for 3h.
- DNase Treatment: Add DNase I, incubate 15 min.
- Purification (TFF): Dilute reaction 5x in PBS, circulate through 30 kDa MWCO cassette. Diafilter against 10 volumes of PBS. Concentrate to ~1 mg/mL.
- Analytics:
- Yield: Quantify by A260 (1 OD = 40 µg/mL).
- Purity: Analyze by capillary electrophoresis (Fragment Analyzer).
- Capping Efficiency: LC-MS/MS for determination of Cap-0, Cap-1, and uncapped species.
- Cost Calculation: Calculate reagent cost per mg of purified, capped mRNA. Compare TFF yield/loss to column-based purification.
Diagram 2: mRNA Vaccine Production & Cost Drivers
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Durability & Stability Research
| Reagent/Kit | Vendor Example | Function in Protocol |
|---|---|---|
| Fluorochrome-Linked Antibodies (anti-mouse CD4, CD19, GL7, CD95) | BioLegend, BD Biosciences | Phenotyping GC B and Tfh cells via flow cytometry (Protocol 2.1). |
| CleanCap Reagent AG (3'-O-Me-m7G) | Trilink Biotechnologies | Co-transcriptional capping for high-fidelity Cap-1 structure in mRNA synthesis (Protocol 4.1). |
| Ribogreen RNA Quantitation Kit | Thermo Fisher Scientific | Ultrasensitive quantification of both encapsulated and free mRNA in LNP formulations. |
| T7 RNA Polymerase (High-Yield) | New England Biolabs, Aldevron | Core enzyme for in vitro transcription of mRNA. Yield optimization is critical for COGs. |
| Sucrose (Pharmaceutical Grade) | MilliporeSigma | Cryoprotectant and lyoprotectant for stabilizing LNPs during lyophilization (Protocol 3.1). |
| Tangential Flow Filtration Cassette (30 kDa MWCO) | Cytiva, Repligen | Scalable, high-recovery purification of large-volume IVT reactions, reducing purification cost. |
| LC-MS/MS Capping Efficiency Assay | e.g., contract research orgs | Gold-standard analytical method to quantify Cap-0, Cap-1, and uncapped mRNA species. |
| SMNP (Saponin/MPLA Nanoparticle) Adjuvant | InvivoGen, custom synthesis | Potent inducer of Th1 and Tfh responses for protein subunit vaccine durability studies. |
Conclusion
The history of viral vaccine development is a story of convergent technological breakthroughs—from empirical observation to cell culture, recombinant genetics, and now programmable nucleic acid platforms. Each paradigm solved critical problems of its era while introducing new optimization challenges. The mRNA/LNP platform represents a fundamental shift from growing or assembling biological pathogens to delivering genetic instructions, offering unparalleled speed and design flexibility. For biomedical researchers, the key takeaway is the acceleration towards a digital-like design-build-test cycle. Future directions will focus on overcoming mRNA's limitations (durability, cost), expanding its application to complex pathogens (HIV, malaria), and integrating AI for antigen design and immune response prediction. The legacy of Jenner to mRNA underscores that the next paradigm will likely hinge on mastering the precise manipulation of adaptive immunity itself, moving from disease-specific vaccines towards programmable immune systems.