LAMP Assay for Rapid Viral Diagnostics: Principles, Applications, and Advanced Protocols for Biomedical Research
This comprehensive review explores Loop-Mediated Isothermal Amplification (LAMP) as a transformative technology for rapid viral detection.
LAMP Assay for Rapid Viral Diagnostics: Principles, Applications, and Advanced Protocols for Biomedical Research
Abstract
This comprehensive review explores Loop-Mediated Isothermal Amplification (LAMP) as a transformative technology for rapid viral detection. Covering foundational principles to advanced applications, we examine LAMP's mechanism leveraging Bst DNA polymerase and multiple primers for isothermal amplification. The article details methodological innovations across diverse pathogens including SARS-CoV-2, MPXV, Ebola virus, and Plasmodium falciparum, while addressing critical troubleshooting aspects like primer design and false-positive reduction. Through comparative validation against gold-standard PCR methods and analysis of clinical performance metrics, we provide researchers and drug development professionals with practical insights for implementing LAMP in both laboratory and point-of-care settings, highlighting its potential to revolutionize diagnostic approaches in infectious disease management.
LAMP Technology Fundamentals: Principles, Mechanisms, and Advantages Over Traditional PCR
Core Principles of Isothermal Amplification and Reaction Mechanics
Loop-mediated isothermal amplification (LAMP) is an advanced nucleic acid amplification technique that enables the rapid, specific, and efficient detection of DNA or RNA targets under constant temperature conditions. First developed by Notomi et al. in 2000, LAMP has emerged as a powerful molecular diagnostic tool that eliminates the requirement for sophisticated thermal cycling equipment, making it particularly valuable for point-of-care testing, field applications, and resource-limited settings [1] [2] [3]. The technique relies on the strand-displacing activity of a specialized DNA polymerase and a unique primer design scheme that targets multiple regions of the nucleic acid sequence of interest [4]. Unlike conventional polymerase chain reaction (PCR), which requires cyclic temperature changes for denaturation, annealing, and extension, LAMP operates isothermally at temperatures between 60-65°C, simplifying instrumentation requirements while maintaining high sensitivity and specificity [4] [2].
The fundamental reaction mechanics of LAMP involve the formation of characteristic loop structures that serve as initiation points for exponential amplification, generating up to 10⁹ copies of the target sequence within 30-60 minutes [1] [2]. This robust amplification capability, combined with versatile detection methods including turbidity, fluorescence, and colorimetric readouts, has positioned LAMP as a transformative technology in diagnostic applications ranging from clinical pathogen detection to agricultural disease monitoring and food safety testing [4] [5] [6]. The core principles governing LAMP technology, including its reaction mechanics, primer design requirements, and detection methodologies, provide the foundation for its expanding applications in rapid viral diagnostics and other molecular detection systems.
Core Reaction Mechanics and Principles
Fundamental Mechanisms of Isothermal Amplification
The LAMP reaction mechanism is characterized by its unique primer design and the formation of self-hybridizing loop structures that enable exponential amplification under isothermal conditions. The process employs a DNA polymerase with high strand displacement activity, typically Bst DNA polymerase derived from Geobacillus stearothermophilus, which can unwind double-stranded DNA without the need for thermal denaturation [1] [2]. The reaction proceeds through a series of precisely orchestrated steps that generate stem-loop DNA structures serving as templates for subsequent amplification cycles.
The initial step involves the binding of inner primers (FIP and BIP) to their complementary target sequences, followed by strand extension. The outer primers (F3 and B3) then bind to adjacent regions and displace the newly synthesized strands through the polymerase's strand displacement activity [2]. These displaced strands form dumbbell-shaped structures due to their self-complementary ends, which serve as the starting material for the cyclic amplification phase [4] [2]. During the cycling amplification step, the inner primers anneal to the loops of the dumbbell structures and initiate successive rounds of strand displacement DNA synthesis, resulting in the formation of long concatemers comprising alternating inverted repeats of the target sequence [1] [2]. This process generates a complex mixture of stem-loop DNAs with various stem lengths and cauliflower-like structures with multiple loops [2].
Table 1: Key Components in LAMP Reaction Mechanics
| Component | Role in Amplification | Key Characteristics |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing enzyme that synthesizes new DNA strands | Lacks 5'→3' exonuclease activity; works at 60-65°C; high processivity [1] |
| Inner Primers (FIP/BIP) | Initiate formation of stem-loop structures | ~40 nt long; contain two distinct binding regions (F1c+F2, B1c+B2) [2] |
| Outer Primers (F3/B3) | Displace synthesized strands to generate loops | ~20 nt long; bind upstream of inner primers [2] |
| Loop Primers (LF/LB) | Accelerate reaction by binding loop regions | Optional but recommended; reduce reaction time by 1/2 to 2/3 [4] |
| Target DNA | Template for amplification | Optimal length: 150-300 bp; minimal size requirement exists [1] |
Primer Design Strategy
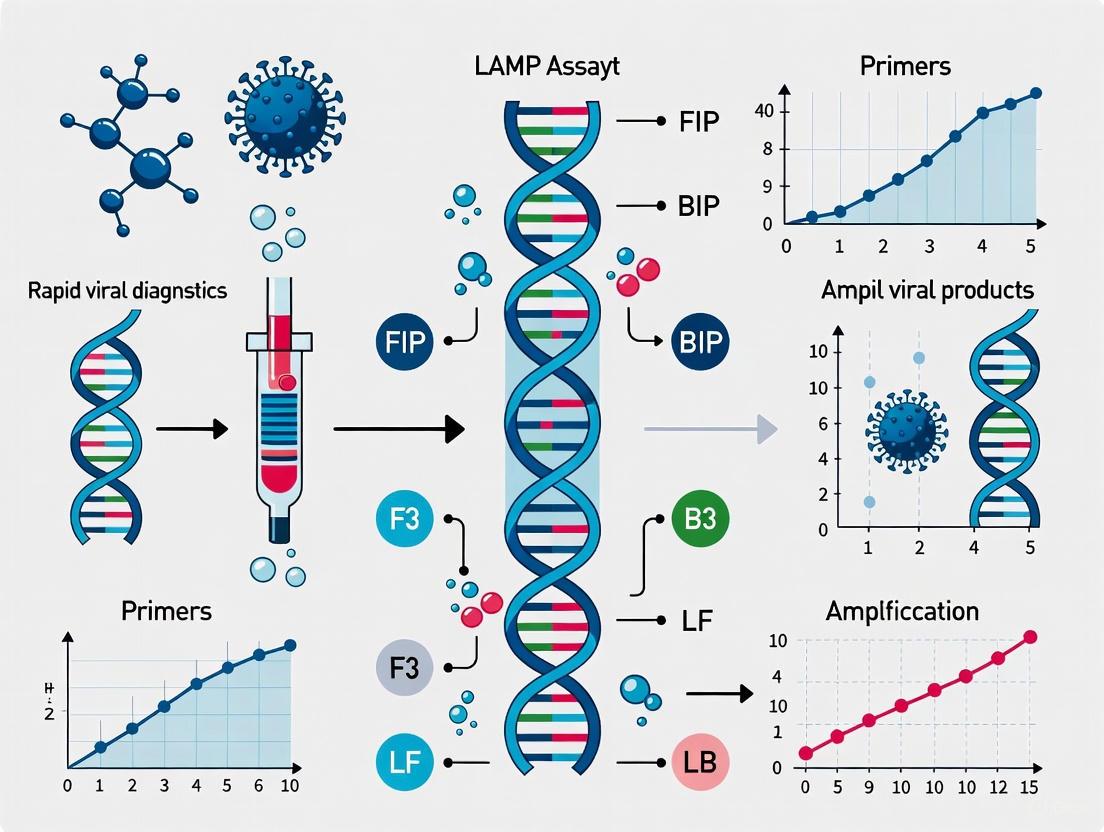
The LAMP primer design strategy is fundamentally different from conventional PCR and represents a critical factor in assay success. A standard LAMP reaction employs four to six primers that recognize six to eight distinct regions on the target gene, providing exceptional specificity [4] [1]. The core primer set consists of two inner primers (Forward Inner Primer - FIP, and Backward Inner Primer - BIP) and two outer primers (F3 and B3) [2]. The FIP contains the F2 region at its 3' end, which is complementary to the F2c region of the target, and the same sequence as the F1c region at its 5' end. Similarly, BIP consists of B2 (complementary to B2c) at the 3' end and B1c at the 5' end [2].
To accelerate the reaction, two additional loop primers (LF and LB) can be incorporated, which bind to the stem-loop structures that are not hybridized by the inner primers [4] [2]. These loop primers prime strand displacement DNA synthesis and can reduce the reaction time by half or more while improving the overall efficiency [4]. The design constraints for LAMP primers are more stringent than for PCR, with specific requirements for the spacing between primer recognition sites, melting temperatures, and GC content [2]. Consequently, specialized software tools such as PrimerExplorer, NEB LAMP Primer Design Tool, and MorphoCatcher are typically employed for primer design rather than manual approaches [4] [3].
Table 2: LAMP Primer Set Composition and Functions
| Primer Type | Recognition Sites | Length | Primary Function |
|---|---|---|---|
| F3 (Forward Outer) | F3c | 17-25 nt | Displaces FIP-linked strand during synthesis |
| B3 (Backward Outer) | B3c | 17-25 nt | Displaces BIP-linked strand during synthesis |
| FIP (Forward Inner) | F2c (3' end) and F1c (5' end) | 40-45 nt | Initiates stem-loop structure formation |
| BIP (Backward Inner) | B2c (3' end) and B1c (5' end) | 40-45 nt | Initiates stem-loop structure formation |
| LF (Loop Forward) | Loop region between F1 and F2 | 17-25 nt | Accelerates reaction by binding single-stranded loop |
| LB (Loop Backward) | Loop region between B1 and B2 | 17-25 nt | Accelerates reaction by binding single-stranded loop |
The following diagram illustrates the LAMP reaction workflow, from initial primer binding to the cyclic amplification phase:
Diagram 1: LAMP Reaction Workflow - illustrating the key stages from initial primer binding to exponential amplification.
Comparative Analysis: LAMP vs. Conventional PCR
LAMP offers distinct advantages and disadvantages compared to conventional PCR, which must be considered when selecting an appropriate amplification method for specific applications. The fundamental differences between these techniques extend beyond their temperature requirements to encompass amplification efficiency, product characteristics, and implementation requirements.
Table 3: Comparative Analysis of LAMP and PCR Characteristics
| Parameter | LAMP | Conventional PCR |
|---|---|---|
| Temperature Requirements | Single temperature (60-65°C) | Multiple temperature cycles (denaturation: 95°C, annealing: 50-65°C, extension: 72°C) [4] |
| Reaction Time | 30-60 minutes [4] [6] | 1.5-2 hours or longer [4] |
| Primer Design | 4-6 primers recognizing 6-8 regions [4] [1] | 2 primers recognizing 2 regions |
| Amplification Efficiency | High (up to 10⁹ copies in <1 hour) [1] | Moderate (typically 10⁶-10⁸ copies in 1.5 hours) |
| Enzyme Used | Bst DNA polymerase (strand-displacing) [1] | Taq DNA polymerase (thermostable) |
| Detection Methods | Turbidity, fluorescence, colorimetric, lateral flow [4] [7] | Gel electrophoresis, fluorescence, sequencing |
| Equipment Needs | Heating block, water bath, or simple thermostat [4] | Thermal cycler required |
| Inhibitor Tolerance | Generally high [4] [3] | Moderate to low |
| Product Type | Long concatemers with cauliflower-like structures [4] | Discrete, single-sized amplicons |
| Quantification Capability | Limited, primarily qualitative [4] | Excellent (especially qPCR) |
| Multiplexing Potential | Challenging due to primer complexity [1] [3] | Well-established |
The isothermal nature of LAMP eliminates the need for expensive thermal cyclers, making it significantly more accessible for field applications and point-of-care testing [4] [3]. The technique's robustness against inhibitors present in complex biological samples allows for minimal sample processing, further enhancing its utility in resource-limited settings [4]. However, LAMP is primarily suited for qualitative detection rather than quantification, and its multiplexing capabilities remain limited compared to PCR due to the complexity of primer design and the heterogeneous nature of amplification products [4] [1]. Additionally, the technique's exceptional sensitivity makes it susceptible to carryover contamination, though this can be mitigated through the incorporation of uracil-DNA-glycosylase (UDG/UNG) and dUTP in the reaction mix [4].
Detection Methods for LAMP Amplicons
LAMP amplification products can be detected through multiple readout systems, ranging from simple visual assessment to sophisticated real-time monitoring instruments. The selection of an appropriate detection method depends on the application requirements, available resources, and desired level of quantification. The most common detection approaches include turbidimetry, fluorometry, colorimetry, and lateral flow assays, each with distinct mechanisms and implementation considerations [4] [7] [1].
Turbidimetry relies on the visual observation or instrumental measurement of increased turbidity resulting from the precipitation of magnesium pyrophosphate, a byproduct of DNA synthesis [1]. This white precipitate becomes visible to the naked eye in positive reactions and can be quantified in real-time using simple photometric equipment [1] [3]. Fluorometry employs DNA-intercalating dyes such as SYTO-9, SYBR Green I, or EvaGreen that fluoresce when bound to double-stranded DNA [1]. This approach enables real-time monitoring of amplification kinetics and can be implemented using portable fluorometers or adapted qPCR instruments [4]. Colorimetric detection utilizes pH-sensitive indicators (e.g., phenol red) or metal ion indicators (e.g., hydroxynaphthol blue, calcein) that undergo visible color changes in response to amplification byproducts [4] [1]. The simplicity of colorimetric readouts makes them particularly suitable for point-of-care applications where equipment availability is limited.
Detailed Detection Protocols
Protocol 1: Colorimetric LAMP Detection Using pH-Sensitive Dyes
Reaction Setup: Prepare LAMP master mix containing 1.5-2.0 mM dNTPs, 6-8 mM MgSO₄, 1× isothermal amplification buffer, 1.6-2.0 µM each of FIP and BIP primers, 0.2-0.4 µM each of F3 and B3 primers, 0.8-1.0 µM each of LF and LB primers (if used), 0.15-0.25 mg/mL phenol red, and 8 U Bst DNA polymerase [4] [8].
Sample Addition: Add 2-5 µL of template DNA to 20-25 µL of master mix. For negative controls, use nuclease-free water instead of template.
Amplification: Incubate reactions at 60-65°C for 30-60 minutes using a heating block, water bath, or portable incubator.
Result Interpretation: Observe color change visually - positive reactions transition from pink (original pH ~8.5) to yellow (acidic pH ~6.0) due to proton release during DNA polymerization [4] [1]. Negative reactions remain pink.
Protocol 2: Fluorescence-Based Real-Time LAMP Detection
Reaction Setup: Prepare LAMP master mix as above, but substitute phenol red with 0.5-1× DNA intercalating dye (SYTO-9, SYBR Green I, or equivalent) [1].
Amplification and Detection: Transfer reactions to appropriate tubes or plates and place in real-time fluorometer or adapted qPCR instrument. Incubate at 60-65°C with fluorescence measurements taken at 1-minute intervals.
Data Analysis: Determine amplification curves and threshold time (Tt) values. Compare Tt values to standard curves for semi-quantitative analysis.
Specificity Verification: Optional melt curve analysis can be performed by gradually increasing temperature from 60°C to 95°C while monitoring fluorescence to confirm specific amplification [4].
Protocol 3: Lateral Flow Detection of LAMP Products
Primer Modification: Design LAMP primers with 5' modifications: FIP or LF primer with biotin, and BIP or LB primer with FAM or FITC [9] [10].
Amplification: Perform standard LAMP reaction with modified primers.
Hybridization and Detection: Dilute amplified product with appropriate buffer and apply to lateral flow strip. As the solution migrates, double-labeled amplicons are captured at the test line by anti-FAM antibodies, while excess biotinylated primers are captured at the control line by streptavidin [9] [10].
Result Interpretation: Positive reactions show both test and control lines, while negative reactions show only the control line.
Table 4: Comparison of LAMP Detection Methods
| Detection Method | Mechanism | Equipment Needs | Sensitivity | Time to Result | Best Applications |
|---|---|---|---|---|---|
| Turbidity | Magnesium pyrophosphate precipitation | Turbidimeter or naked eye | Moderate | Endpoint or real-time | Field testing with basic equipment [1] |
| Fluorescence | DNA-intercalating dyes | Fluorometer or qPCR instrument | High | Real-time | Laboratory quantification [4] [1] |
| Colorimetric (pH) | Proton release during polymerization | Naked eye | Moderate | Endpoint | Point-of-care, resource-limited settings [4] [1] |
| Colorimetric (Metal Ion) | Mg²⁺ depletion | Naked eye | Moderate | Endpoint | Field applications [1] |
| Lateral Flow | Immunochromatography | Naked eye | High | Endpoint (post-amplification) | Point-of-care, home testing [9] [10] |
The following diagram illustrates the relationships between different LAMP detection methods and their appropriate applications:
Diagram 2: LAMP Detection Methods and Applications - showing the relationships between detection techniques and their primary implementation contexts.
Research Reagent Solutions and Essential Materials
Successful implementation of LAMP assays requires careful selection of reagents and materials optimized for isothermal amplification. The following table outlines key components and their functions in standardized LAMP reactions:
Table 5: Essential Research Reagents for LAMP Assays
| Reagent/Material | Function | Recommended Specifications | Alternative Options |
|---|---|---|---|
| Bst DNA Polymerase | Strand-displacing enzyme for isothermal amplification | Bst 2.0 WarmStart or Bst 3.0 (with reverse transcriptase activity) [1] | OmniAmp polymerase, Bst LF fragment |
| Primer Sets | Target recognition and amplification initiation | HPLC-purified, 4-6 primers per target [4] | Custom-designed using NEB tool or PrimerExplorer |
| dNTPs | Building blocks for DNA synthesis | 1.5-2.0 mM final concentration, molecular biology grade | dNTP mix including dUTP for carryover prevention [4] |
| Magnesium Ions | Cofactor for polymerase activity and pyrophosphate precipitation | 6-8 mM MgSO₄ [4] | MgCl₂ (concentration may need optimization) |
| Betaine | Stabilizer for strand separation | 0.8-1.0 M final concentration [9] | Trimethylglycine, DMSO (optional) |
| Detection Dye | Amplification visualization | Phenol red (colorimetric), SYTO-9 (fluorescence) [4] [1] | Hydroxynaphthol blue, calcein, SYBR Green I |
| Uracil-DNA Glycosylase (UDG/UNG) | Carryover contamination prevention | Thermolabile UDG for single-tube applications [4] | Standard UNG with heat inactivation step |
| Reverse Transcriptase | RNA template conversion (RT-LAMP) | MMLV RT, Bst 3.0 (with intrinsic RT activity) [1] | AMV RT (higher temperature compatibility) |
Applications in Viral Diagnostics and Future Perspectives
LAMP technology has demonstrated significant utility in viral diagnostics, particularly in scenarios requiring rapid results, minimal equipment, and high sensitivity. The COVID-19 pandemic highlighted the value of LAMP-based approaches, with RT-LAMP assays developed for SARS-CoV-2 detection that provided results within 30-40 minutes without sophisticated instrumentation [4] [3]. Similar applications have been reported for various viral pathogens including Zika virus, hepatitis viruses, and influenza [5] [3]. The technique's robustness against inhibitors has enabled its implementation with minimally processed samples such as saliva, nasopharyngeal swabs, and blood, further streamlining the diagnostic workflow [4] [3].
Recent advancements in LAMP technology have focused on enhancing speed, specificity, and integration with complementary technologies. Engineering of improved Bst polymerase variants (Bst 2.0, Bst 3.0) has resulted in enzymes with superior polymerization speed, thermal stability, and reverse transcriptase activity [1]. Integration of LAMP with CRISPR-Cas systems has enabled highly specific detection through collateral cleavage activity, while microfluidic platforms have facilitated the development of automated, high-throughput LAMP systems [1]. These technological innovations continue to expand the application landscape for LAMP, particularly in point-of-care testing, environmental monitoring, and food safety assurance.
Future developments in LAMP technology are likely to focus on multiplexing capabilities, quantitative analysis, and further simplification of sample processing steps. The combination of LAMP with portable electronic readers and smartphone-based detection platforms represents a promising direction for democratizing molecular diagnostics [5]. Additionally, the integration of LAMP with sample preparation technologies in lab-on-a-chip formats may enable complete sample-to-answer systems for field deployment. As these advancements continue to mature, LAMP is poised to play an increasingly important role in global health security, agricultural biosecurity, and personalized medicine applications.
Loop-mediated isothermal amplification (LAMP) has emerged as a transformative technology in molecular diagnostics, particularly for rapid viral detection. This application note details the core components of the LAMP assay system, focusing on the engineered Bst DNA polymerase and the sophisticated multi-primer architecture that underpin its exceptional performance. The isothermal nature of LAMP, requiring only a simple heating block or water bath, provides significant advantages over traditional PCR in resource-limited settings and point-of-care diagnostics [11] [12]. The robustness of this system enables detection of viral pathogens with sensitivity down to 20 copies/µL and results in less than 30 minutes, making it invaluable for rapid response in outbreak situations [13].
Core Principles of LAMP Technology
LAMP is an autocatalytic DNA amplification process that occurs under isothermal conditions (typically 60-65°C) through the combined activity of a strand-displacing DNA polymerase and a set of specifically designed primers [14] [12]. Unlike PCR, which requires thermal cycling between denaturation, annealing, and extension temperatures, LAMP accomplishes exponential amplification at a single temperature by leveraging the inherent strand displacement activity of Bst DNA polymerase [12]. This fundamental difference eliminates the need for expensive thermocyclers and simplifies the instrumentation required for molecular diagnostics.
The amplification mechanism produces long DNA concatemers (>20 kb) consisting of numerous repeats of the target sequence connected by single-stranded loop regions [11]. These products can be detected through multiple methods including real-time fluorescence, colorimetric change, turbidity, or lateral flow detection, providing flexibility for different diagnostic settings and applications [11] [14].
Bst DNA Polymerase: The Engine of LAMP Amplification
Biochemical Properties and Engineering
Bst DNA polymerase, derived from the large fragment of Geobacillus stearothermophilus DNA Polymerase I, serves as the core enzyme in LAMP reactions due to its robust strand displacement activity and absence of 5'→3' exonuclease activity [15]. The enzyme operates optimally at 60-65°C and can amplify target DNA in as little as 10-15 minutes under ideal conditions [12]. The large fragment (approximately 68 kDa) retains the polymerase functionality while lacking the exonuclease domain that would otherwise interfere with strand displacement amplification [15].
Recent protein engineering efforts have significantly enhanced Bst DNA polymerase performance through strategic modifications:
- Charge engineering: Adding multiple charged residues to surface domains improves thermostability and diagnostic performance, enabling LAMP at temperatures up to 74°C [16]
- Domain fusion: Incorporation of DNA-binding domains such as Helix-hairpin-helix (HhH) motifs enhances processivity and salt tolerance [15]
- Reverse transcriptase activity: Engineered variants (e.g., Bst 3.0) enable single-enzyme RT-LAMP reactions for direct RNA detection [13]
Table 1: Comparison of Bst DNA Polymerase Variants
| Polymerase Variant | Optimal Temperature | Key Features | Applications |
|---|---|---|---|
| Bst 2.0 | 65°C | High strand displacement, warm-start capability | Standard LAMP assays |
| Bst 3.0 | 65°C | Built-in reverse transcriptase activity | Single-enzyme RT-LAMP |
| Bst-XT | 65°C | Combines specificity of Bst 2.0 with speed of Bst 3.0 | Rapid diagnostics (<15 min) |
| Engineered Br512 | Up to 74°C | Charge-enhanced thermostability | High-temperature LAMP, inhibitor-tolerant assays |
Reaction Components and Optimization
The LAMP reaction mixture contains several key components that must be optimized for robust performance:
- Bst DNA polymerase (typically 0.2-0.4 µM) with strand displacement activity [16]
- dNTPs (1.4 mM) as DNA building blocks [16]
- Magnesium ions (8 mM MgCl₂) as essential cofactors [16]
- Betaine (0.4 M) to enhance strand separation and prevent secondary structure formation [16]
- Primer mix (inner, outer, and loop primers) targeting multiple regions of the DNA template [13]
Reaction optimization often includes the addition of guanidine hydrochloride (40 mM), which has been shown to improve detection speed by 22% and enhance overall assay efficiency [13].
Multi-Primer System Architecture
Primer Design Strategy
The LAMP primer system represents a sophisticated architectural design that targets multiple distinct regions of the target DNA sequence. A complete primer set typically includes:
- Forward Inner Primer (FIP): Comprising F2 region (at the 3' end) and F1c region (at the 5' end)
- Backward Inner Primer (BIP): Comprising B2 region (at the 3' end) and B1c region (at the 5' end)
- Forward Outer Primer (F3): Binding upstream of FIP
- Backward Outer Primer (B3): Binding upstream of BIP
- Loop Forward Primer (LF): Accelerating reaction by binding loop regions
- Loop Backward Primer (LB): Accelerating reaction by binding loop regions
These primers typically range from 15-25 bases in length with a GC content of 40-60%, and should avoid runs of 3 or more identical nucleotides or dinucleotide repeats [17]. The outer primers (F3/B3) generally have Tm values of 55-63°C, while inner and loop primers have higher Tm values (60-68°C), with maximum differences of 5°C between primer pairs [17].
Five-Primer vs. Six-Primer Systems
Recent research has demonstrated that five-primer LAMP systems (omitting one loop primer) can significantly reduce false-positive rates while maintaining high sensitivity [13]. In comparative studies, a five-primer E-ID1 set targeting the SARS-CoV-2 E gene showed no misamplification even after 120 minutes, whereas six-primer sets began showing false positives in as little as 40 minutes [13]. The five-primer system achieved sensitivity of 89.5% (colorimetric) and 92.2% (fluorometric) with a limit of detection of 20 copies/µL, demonstrating that careful primer selection can optimize the trade-off between speed and specificity [13].
Table 2: Performance Comparison of Primer Systems
| Parameter | Five-Primer System | Six-Primer System |
|---|---|---|
| False Positive Rate | Significant reduction (no misamplification after 120 min) | Higher (misanplification in 40 min) |
| Sensitivity | 89.5-92.2% | Typically higher (95-98%) |
| Amplification Time | 27-30 min (with optimization) | 15-20 min |
| Specificity | 97.2-99% | Slightly lower due to primer interactions |
| Recommended Use | Clinical diagnostics requiring high specificity | Rapid screening where speed is critical |
Advanced LAMP Methodologies
Multiplex LAMP Strategies
Multiplexing LAMP assays to detect multiple targets simultaneously presents significant technical challenges due to the complexity of primer interactions, but several innovative approaches have been developed:
- DARQ-LAMP: Uses quencher-labeled primers and fluorophore-labeled complementary sequences that separate during amplification, enabling real-time detection of up to four targets [18]
- QUASR-LAMP: Employs fluorophore-labeled primers and quencher-labeled probes that remain separate during amplification but hybridize upon cooling for endpoint detection [18]
- Multiple Endonuclease Restriction Real-Time LAMP: Incorporates restriction enzyme recognition sites into primers, with cleavage during amplification generating fluorescence signal [19]
- Target-Specific Fluorogenic Probes: Uses sequence-specific probes with different fluorophores to distinguish multiple targets in a single reaction [18]
Each multiplexing methodology offers distinct advantages in real-time detection capability, ease of result interpretation, compatibility with point-of-care use, and maximum target number, with the choice depending on specific application requirements [18].
Detection Methodologies
The extensive amplification in LAMP reactions (producing micrograms of DNA) enables detection through multiple direct and indirect methods:
Complete Experimental Protocol
RT-LAMP for RNA Virus Detection
Principle: This protocol describes a one-step reverse transcription LAMP (RT-LAMP) assay for detection of RNA viruses (e.g., SARS-CoV-2) using either Bst 2.0 + separate reverse transcriptase or Bst 3.0 with intrinsic reverse transcriptase activity [13].
Reagents and Equipment:
- Bst 2.0 or Bst 3.0 DNA polymerase (commercial LAMP master mixes available)
- dNTP mix (1.4 mM final concentration)
- LAMP primers (FIP/BIP: 2.4 µM each; F3/B3: 0.6 µM each; LF/LB: 1.2 µM each)
- Betaine (0.4 M final concentration)
- Magnesium sulfate (8 mM final concentration)
- Guanidine hydrochloride (40 mM final concentration, optional for enhanced performance)
- WarmStart mechanism to prevent non-specific amplification
- Heating block or water bath (65°C)
- Microcentrifuge tubes or 96-well plate
Procedure:
- Reaction Setup (on ice):
- Prepare reaction mix (25 µL total volume):
- 12.5 µL 2X LAMP master mix
- 1.0 µL primer mix (containing all LAMP primers)
- 1.0 µL GuHCl (40 mM stock, optional)
- 5.5 µL nuclease-free water
- 5.0 µL RNA template
- Mix gently by pipetting, avoid bubbles
- Centrifuge briefly to collect reaction at tube bottom
- Prepare reaction mix (25 µL total volume):
Amplification:
- Place reactions in preheated thermal block at 65°C
- Incubate for 30-60 minutes
- For real-time monitoring, take fluorescence readings every 3 minutes
Detection:
- Colorimetric: Visual inspection for color change from pink to yellow
- Fluorometric: Measure fluorescence signal (FAM channel for common probes)
- Endpoint confirmation: 2% agarose gel electrophoresis for ladder pattern
Reaction Termination:
- Heat to 80°C for 2 minutes to stop reaction
- Analyze results immediately or store at 4°C
Troubleshooting Guide:
- No amplification: Check primer design, enzyme activity, and template quality
- False positives: Include UDG treatment for carryover prevention, use WarmStart enzymes
- Late amplification: Optimize Mg²⁺ concentration, add GuHCl, increase enzyme amount
- Non-specific bands: Redesign primers, increase reaction temperature, optimize primer ratios
Multiplex LAMP Using DARQ Methodology
Principle: This protocol enables simultaneous detection of multiple targets in a single reaction using Detection of Amplification by Release of Quenching (DARQ) [18].
Reagents:
- Standard LAMP reagents as in protocol 6.1
- Quencher-labeled FIP primers (5' end)
- Fluorophore-labeled complementary sequences
- Multiple fluorophores with distinct emission spectra (FAM, HEX, Cy5, etc.)
Procedure:
- QPD Probe Preparation:
- Design FIP primer with 5' quencher and complementary sequence with 3' fluorophore
- Anneal quencher-primer and fluorophore-probe to form Quencher Probe Duplex (QPD)
- Incorporate QPD into LAMP reaction mix
Multiplex Reaction Setup:
- Prepare separate primer sets for each target with distinct fluorophores
- Use 100 nM final concentration of each QPD probe
- Keep total primer concentration balanced to avoid inhibition
Amplification and Detection:
- Perform LAMP at 65°C with real-time fluorescence monitoring
- Monitor multiple fluorescence channels simultaneously
- As amplification proceeds, fluorophore is released from quencher, generating target-specific signal
Research Reagent Solutions
Table 3: Essential Reagents for LAMP Assay Development
| Reagent Category | Specific Examples | Function | Optimization Notes |
|---|---|---|---|
| DNA Polymerases | Bst 2.0, Bst 3.0, Bst-XT, Bsm | Strand displacement amplification | Bst 3.0 includes reverse transcriptase; Bst-XT offers speed + specificity |
| Primer Design Tools | NEB LAMP Primer Design Tool, PrimerExplorer, GLAPD | In silico primer design | Automated tools handle complex design constraints; validate empirically |
| Detection Chemistries | Hydroxynaphthol blue, calcein, SYTO dyes, WarmStart Colorimetric Master Mix | Amplification visualization | Colorimetric master mixes include pH-sensitive dyes for direct visualization |
| Reverse Transcriptases | WarmStart RTx, M-MuLV | RNA template conversion | Not needed with Bst 3.0; required for other Bst variants in RT-LAMP |
| Reaction Enhancers | Betaine, guanidine hydrochloride, DMSO | Improve efficiency and specificity | GuHCl reduces detection time by 22%; optimize concentration for each assay |
| Carryover Prevention | UDG treatment, dUTP incorporation | Contamination control | WarmStart Colorimetric LAMP Master Mix with UDG prevents false positives |
Applications in Viral Diagnostics
LAMP technology has demonstrated particular utility in rapid viral diagnostics across diverse applications:
- SARS-CoV-2 detection: Five-primer E-ID1 RT-LAMP achieved 97.2% specificity and 94.5% accuracy compared to RT-PCR [13]
- Influenza and respiratory viruses: Multiplex RT-LAMP enables differential diagnosis of co-circulating pathogens [14]
- Hepatitis viruses: LAMP assays developed for both HBV (DNA) and HCV (RNA) detection with sensitivity comparable to PCR [14]
- Emerging arboviruses: Rapid detection of Dengue, Zika, and Chikungunya viruses in field settings [14]
- Veterinary diagnostics: Capripox virus detection with 100% sensitivity for GTPV and 98.8% for SPPV [20]
The robustness of LAMP assays to inhibitors present in clinical samples (blood, saliva, tissue) enables minimal processing and rapid time-to-result, making it particularly valuable for point-of-care applications and resource-limited settings [11] [20].
The synergistic combination of engineered Bst DNA polymerase variants and sophisticated multi-primer system architecture has established LAMP as a powerful technology platform for rapid molecular diagnostics. Ongoing advancements in enzyme engineering, primer design strategies, multiplexing methodologies, and detection technologies continue to expand the applications and performance boundaries of LAMP assays. The five-primer approach represents a significant innovation in addressing the traditional limitation of false positives while maintaining high sensitivity. As these technologies mature, LAMP-based diagnostics are poised to play an increasingly important role in global health security, outbreak response, and point-of-care testing across diverse healthcare settings.
Loop-mediated isothermal amplification (LAMP) represents a paradigm shift in nucleic acid amplification technology, distinguished by its unique structural mechanism that recognizes six to eight distinct sequences on target DNA or RNA. This application note details the underlying molecular basis for LAMP's exceptional specificity and provides detailed protocols for implementing this technology in viral diagnostics. Compared to conventional PCR that utilizes only two primers recognizing a single sequence, LAMP's multi-primer architecture enables unmatched selectivity, making it particularly valuable for rapid pathogen detection in point-of-care settings. We present comprehensive experimental workflows, optimized reagent formulations, and validation data to support researchers in deploying this powerful methodology for viral diagnostics and therapeutic development.
The exceptional specificity of loop-mediated isothermal amplification (LAMP) stems directly from its unique structural design employing multiple primers that recognize an extensive array of target sites. Where conventional PCR relies on just two primers binding to a single target sequence, the LAMP mechanism utilizes four to six primers that collectively identify six to eight distinct regions on the target genome [21] [22]. This multi-primer architecture creates a structural safeguard that virtually eliminates false positives from non-specific amplification.
The significance of this structural advantage extends throughout the diagnostic pipeline. For viral detection, LAMP's requirement for simultaneous recognition of multiple conserved regions provides built-in protection against amplification of non-target sequences or related viral strains with partial homology. This intrinsic specificity allows LAMP to maintain excellent performance even when using simplified sample preparation methods that may contain inhibitory substances [10]. The structural foundation of LAMP therefore enables both high precision and practical utility in field-based settings where rapid viral diagnostics are most critical.
Mechanism: Multi-Primer Recognition System
Core Primer Architecture
The LAMP system employs a sophisticated primer design that facilitates the recognition of multiple target sites:
Inner Primers (FIP/BIP): These form the core of the amplification mechanism, with each inner primer containing two distinct recognition sequences (F1c+F2 for FIP; B1c+B2 for BIP) that target complementary regions on the sense and antisense strands of the target DNA [21]. These extended primers (typically 45-49 bp) initiate the formation of the characteristic loop structures.
Outer Primers (F3/B3): Shorter primers (21-24 bp) that bind at positions flanking the inner primer recognition sites. Their primary function is to displace synthesized strands during the amplification process, facilitating strand displacement DNA synthesis [21] [22].
Loop Primers (LF/LB): Optional but highly beneficial primers that recognize the loop structures formed during later amplification stages. By binding to these structures, loop primers accelerate reaction times by up to 50% and increase the total number of recognition sites to eight distinct sequences [22].
Structural Mechanism Workflow
The following diagram illustrates the coordinated interaction of LAMP primers with their multiple target recognition sites:
Figure 1: LAMP primer system showing six to eight target recognition sites that provide structural basis for high specificity
The molecular mechanism proceeds through three distinct phases:
Initial Amplification: The forward inner primer (FIP) binds to the target DNA and initiates complementary strand synthesis. The outer primer (F3) then binds and initiates strand displacement, releasing a single-stranded DNA molecule that serves as template for subsequent priming by the backward primers (BIP and B3) [21].
Loop Formation and Cycling Amplification: The released single-stranded DNA forms a stem-loop structure at each end due to complementary F1c/F1 and B1c/B1 regions. This dumbbell-shaped structure serves as the starting material for exponential amplification, where inner primers continuously prime on the loop structures [21] [22].
Exponential Amplification: The cycling reaction continues with accumulation of up to 10⁹ copies of target in less than an hour, producing stem-loop DNAs with several inverted repeats that form complex cauliflower-like structures with multiple loops [21].
Research Reagent Solutions
Table 1: Essential research reagents for LAMP assay development and implementation
| Reagent Category | Specific Examples | Function & Importance in LAMP |
|---|---|---|
| DNA Polymerase | Bst DNA polymerase large fragment (New England Biolabs) [21], Bst 2.0 WarmStart, Bst 3.0 [22] | Strand-displacing activity essential for isothermal amplification; thermostable variants maintain activity at 60-65°C |
| Primer Sets | FIP/BIP, F3/B3, LF/LB [21] [23] | Core recognition system; 4-6 primers targeting 6-8 regions provide structural specificity |
| Buffer Components | Betaine (0.8-1.6 M) [21], MgSO₄ (4-8 mM) [23], dNTPs (1.0-1.4 mM) [23], Tris-HCl (pH 8.8) | Betaine reduces secondary structure in GC-rich regions; magnesium is essential cofactor for polymerase activity |
| Detection Systems | SYBR Green I [24], Hydroxynaphthol Blue [23], colorimetric pH indicators [25], calcein [22] | Enable visual detection of amplification; color change or fluorescence indicates positive reaction |
| Reverse Transcriptase | RTx (New England Biolabs) [25] | Essential for RT-LAMP applications for RNA virus detection; may be combined with DNA polymerase in master mixes |
Experimental Protocols
Standard LAMP Reaction Protocol
The following optimized protocol is adapted from multiple established LAMP assays for viral detection [21] [26] [23]:
Table 2: Standard LAMP reaction components and conditions
| Component | Final Concentration | Volume (25 µl reaction) | Notes |
|---|---|---|---|
| Reaction Buffer | 1X | 12.5 µl | 20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH₄)₂SO₄, 2 mM MgSO₄, 0.1% Tween 20 |
| MgSO₄ | 4-8 mM | 1-2 µl (from 100 mM stock) | Optimize for each primer set; critical for polymerase activity |
| dNTPs | 1.2-1.4 mM | 2-2.5 µl (from 10 mM stock) | Equal mixture of dATP, dCTP, dGTP, dTTP |
| Betaine | 0.8-1.6 M | 3-4 µl (from 5 M stock) | Reduces secondary structure; essential for GC-rich targets |
| Primers | Variable | 1-2 µl total | See Table 3 for specific concentrations |
| Bst DNA Polymerase | 8-12 U | 0.5-1 µl | Strand-displacing polymerase (e.g., Bst 2.0 WarmStart) |
| Template DNA | 1-50 ng | 2-5 µl | Can use purified DNA or crude lysates |
| Nuclease-free Water | - | To 25 µl | - |
Primer Concentration Optimization: Table 3: Recommended primer concentrations for LAMP assays
| Primer Type | Final Concentration | Function |
|---|---|---|
| FIP/BIP | 0.8-1.6 µM each [23] | Inner primers initiating stem-loop formation |
| F3/B3 | 0.1-0.2 µM each [21] [23] | Outer primers enabling strand displacement |
| LF/LB | 0.2-0.4 µM each [23] | Loop primers accelerating reaction speed |
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 2-5 minutes (optional for GC-rich targets) [21]
- Isothermal Amplification: 60-65°C for 15-60 minutes [23]
- Enzyme Inactivation: 80°C for 5-10 minutes [21]
Specificity Validation Protocol
To confirm the structural specificity of LAMP primers, implement the following validation workflow:
Figure 2: Comprehensive workflow for validating LAMP assay specificity
Implementation Notes:
- Inclusivity Testing: Include 5-10 different strains of the target virus representing genetic diversity [24]
- Exclusivity Testing: Test against 10-20 near-neighbor species or common co-infecting pathogens [24]
- Clinical Samples: Validate with at least 50 positive and 100 negative clinical samples for statistical significance [25]
Detection and Visualization Methods
Table 4: LAMP product detection methods with applications and sensitivity
| Detection Method | Principle | Application Context | Sensitivity | Equipment Needed |
|---|---|---|---|---|
| Colorimetric (pH indicator) | pH change due to pyrophosphate release [25] | Point-of-care testing, field use | Visible at >10⁹ copies | None (naked eye) |
| Fluorescent Dyes | SYBR Green I, intercalating dyes [24] [23] | Laboratory setting, quantitative analysis | 100 fg DNA [24] | UV light or blue light illuminator |
| Turbidity | Magnesium pyrophosphate precipitation [22] | Resource-limited settings | Visual turbidity at >0.5 mM | None (naked eye) |
| Lateral Flow Dipstick | Biotin-labeled primers with immunochromatography [10] | Field deployment, multiplex detection | Comparable to fluorescence | None (dipstick) |
| Real-time Monitoring | Continuous fluorescence measurement [23] | Quantitative applications, kinetics | 10 copies/reaction [26] | Real-time isothermal instrument |
Performance Data and Applications
Sensitivity and Specificity Metrics
Table 5: Performance comparison of LAMP assays for pathogen detection
| Target Pathogen | Detection Limit | Specificity | Time to Result | Reference |
|---|---|---|---|---|
| Dickeya fangzhongdai (plant pathogen) | 100 fg (18-20 genome copies) [24] | 100% (96/96 strains) [24] | <60 minutes [24] | [24] |
| SARS-CoV-2 | 10 copies per reaction [26] | 99.7% vs RT-qPCR [25] | 40 minutes [26] | [26] [25] |
| Plasmopara halstedii (sunflower pathogen) | 0.5 pg/μl [23] | Specific to target species [23] | 45 minutes [23] | [23] |
| Diarrheagenic E. coli | 10²–10³ gene copies/reaction [10] | Moderate specificity for eae and stx2 genes [10] | 30-45 minutes [10] | [10] |
Applications in Viral Diagnostics
The structural specificity of LAMP enables several critical applications in viral diagnostics:
Point-of-Care Testing: The ability to maintain specificity with minimal equipment makes LAMP ideal for field deployment. The COVID-19 pandemic demonstrated RT-LAMP's utility for large-scale screening with sensitivity of 97.5% and specificity of 99.7% compared to RT-qPCR for samples with Ct values <30 [25].
Emerging Variant Detection: Properly designed LAMP assays targeting conserved regions can detect emerging viral variants. One SARS-CoV-2 LAMP assay targeting the N gene (positions 12-213) maintained robust detection capability across variants of concern including Alpha, Beta, Delta, and Omicron [26].
Multiplex Detection: The incorporation of multiple primer sets enables simultaneous detection of several pathogens in a single reaction. Molecular beacon probes and nucleic acid lateral flow detection allow for discrimination between multiple targets in duplex LAMP assays [10].
Troubleshooting Guide
Table 6: Common LAMP challenges and solutions
| Problem | Potential Causes | Solutions |
|---|---|---|
| No amplification | Primer design issues, insufficient Mg²⁺, enzyme inhibition | Verify primer specificity in silico, optimize Mg²⁺ concentration (4-12 mM), use internal control |
| Non-specific amplification | Primer-dimer formation, low annealing stringency | Redesign primers with stricter criteria, increase temperature (65°C), use WarmStart enzymes |
| Late amplification | Suboptimal primer concentrations, insufficient enzyme | Increase inner primer concentration (up to 1.6 µM), add loop primers, increase Bst polymerase (8-12 U) |
| Inconsistent results | Template impurities, inhibitor carryover | Implement sample purification, add betaine (1 M), include positive and negative controls |
The structural basis of LAMP technology—specifically its utilization of six to eight target recognition sites—provides an unparalleled foundation for highly specific molecular diagnostics. This multi-primer recognition system creates a built-in verification mechanism that significantly reduces false positives compared to conventional two-primer amplification methods. The protocols and data presented herein demonstrate that properly designed LAMP assays can achieve sensitivity down to single-digit copy numbers while maintaining exceptional specificity across diverse viral targets.
For researchers developing viral diagnostic tests, LAMP offers a compelling combination of precision, speed, and practical deployability. The technology's compatibility with simple visualization methods and minimal equipment requirements positions it as an ideal platform for both laboratory-based testing and point-of-care applications. As viral threats continue to emerge, the structural specificity of LAMP ensures it will remain a valuable tool for rapid response and containment efforts.
Loop-mediated isothermal amplification (LAMP) has emerged as a transformative nucleic acid amplification technique that addresses critical limitations of traditional reverse transcription quantitative polymerase chain reaction (RT-qPCR) in viral diagnostics. While RT-qPCR remains the gold standard for detecting viral pathogens like SARS-CoV-2, its dependency on sophisticated thermal cycling instrumentation, lengthy processing times, and complex laboratory infrastructure has stimulated the search for alternative methodologies suitable for point-of-care testing and resource-limited settings [27] [28]. LAMP technology, first developed by Notomi et al. in 2000, enables rapid nucleic acid amplification at a constant temperature through the use of strand-displacing DNA polymerase and multiple primers recognizing distinct regions of the target sequence [29]. This technical note provides a comparative analysis of LAMP versus RT-qPCR, focusing on the core advantages of speed, simplicity, and reduced equipment requirements, supported by experimental data and detailed protocols relevant to researchers and drug development professionals working in viral diagnostics.
Technical Comparison: LAMP vs. RT-qPCR
Fundamental Technological Differences
The fundamental distinction between these methodologies lies in their amplification mechanisms. RT-qPCR requires thermal cycling between precise temperatures for denaturation, annealing, and extension, typically involving 35-45 cycles over 1-2 hours [28]. In contrast, LAMP employs isothermal amplification at 60-65°C using 4-6 primers that recognize 6-8 distinct regions of the target gene, generating up to 10⁹ copies in under an hour through a complex process involving strand displacement and the formation of loop structures [22] [29]. This eliminates the need for precise thermal cycling and enables amplification with simpler equipment.
Speed and Throughput Comparison
Multiple studies have demonstrated the significant time reduction offered by LAMP compared to RT-qPCR. A 2024 evaluation reported that RT-LAMP assays could provide results within 30-45 minutes, substantially faster than the several hours typically required for RT-qPCR including RNA extraction and amplification [30] [28]. This rapid detection is particularly valuable during outbreak situations where timely diagnosis is critical for infection control and prompt treatment initiation.
Table 1: Time Efficiency Comparison Between RT-LAMP and RT-qPCR
| Process Step | RT-LAMP | RT-qPCR |
|---|---|---|
| Sample Preparation | 10-30 minutes [31] | 30-60 minutes [28] |
| Amplification Time | 15-45 minutes [30] [32] | 1-2 hours [28] |
| Total Time to Result | 30-60 minutes | 2-4 hours |
| Hands-on Time | Minimal [31] | Significant [28] |
Equipment and Infrastructure Requirements
The equipment requirements for LAMP are substantially less complex than those for RT-qPCR, making LAMP more suitable for resource-limited settings and point-of-care applications. While RT-qPCR requires expensive thermal cyclers costing thousands of dollars, LAMP reactions can be performed using simple dry bath heaters, water baths, or portable incubators maintaining a single temperature [22] [31]. Furthermore, result interpretation for LAMP does not necessarily require sophisticated detection systems; positive amplification can be visualized through colorimetric changes, turbidity, or fluorescence using portable readers or even the naked eye [22] [33].
Table 2: Equipment Requirements Comparison
| Equipment Type | RT-LAMP | RT-qPCR |
|---|---|---|
| Amplification Device | Dry bath, water bath, simple incubator [22] | Sophisticated thermal cycler [28] |
| Detection System | Naked eye, portable reader, smartphone [31] [33] | Fluorescence detection system [30] |
| Cost | Low | High |
| Portability | High [31] | Low |
| Power Requirements | Simple | Complex |
Sensitivity and Specificity Performance
When properly optimized, RT-LAMP demonstrates comparable diagnostic accuracy to RT-qPCR during the acute phase of infection. A 2021 study examining 124 nasopharyngeal samples from COVID-19 patients reported that RT-LAMP maintained a positivity of 92.8% and 100% sensitivity and specificity compared to RT-qPCR up to the 9th day after symptom onset [32]. The limit of detection for optimally designed RT-LAMP assays has been reported as low as 6.7 copies per reaction [32] or 50 RNA copies per μL in viral transport medium [31]. The high specificity of LAMP stems from its use of multiple primers (typically 4-6) that recognize 6-8 distinct regions of the target sequence, making false positives due to non-specific amplification less likely than with traditional PCR methods [22].
Experimental Protocols
One-Step RT-LAMP Reaction Protocol
The following protocol has been adapted from published methodologies for SARS-CoV-2 detection [30] [33] and can be modified for other viral targets through appropriate primer design.
Reagent Preparation
- Prepare a 25 μL reaction mixture containing:
- 12.5 μL of 2× LAMP reaction mix
- 1.0 μL of Bst DNA/RNA Polymerase (8 U/μL)
- 5 pmol each of F3 and B3 external primers
- 40 pmol each of FIP and BIP internal primers
- 20 pmol each of LF and LB loop primers
- 5 μL of RNA template
- Nuclease-free water to 25 μL
Note: Primer design is critical for successful LAMP amplification. Use specialized software such as PrimerExplorer V5 for designing primers that recognize 6 distinct regions of the target sequence.
Amplification Conditions
- Incubate the reaction mixture at 65°C for 30-45 minutes
- Terminate the reaction by heating at 80°C for 5 minutes to inactivate the enzyme
Result Interpretation
Several detection methods can be employed:
- Colorimetric: Positive reaction changes from pink to yellow [33]
- Turbidimetric: Increased turbidity due to magnesium pyrophosphate precipitate [22]
- Fluorometric: Fluorescence increase with intercalating dyes [31]
- Visual: Green fluorescence under UV light [28]
Portable Point-of-Care Detection Protocol
For field applications, a smartphone-based detection system can be implemented as follows [31]:
- Sample Collection: Collect nasopharyngeal swab and transfer to viral transport medium
- Heat Lysis: Incubate aliquot at 95°C for 1 minute
- Cartridge Loading: Transfer lysed sample and LAMP reagents to microfluidic cartridge
- Isothermal Amplification: Incubate at 65°C for 30 minutes in portable heater
- Smartphone Detection: Monitor fluorescence in real-time using smartphone camera and dedicated cradle
- Data Analysis: Use mobile application for result interpretation and threshold time calculation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for LAMP-Based Viral Detection
| Reagent/Component | Function | Examples & Specifications |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase for isothermal amplification | Bst 2.0, Bst 3.0 (with reverse transcriptase activity) [29] |
| Primer Sets | Recognize multiple target regions for specific amplification | 4-6 primers targeting 6-8 regions; designed via PrimerExplorer [30] |
| Detection Dyes | Visual or fluorescent signal generation | Hydroxy naphthol blue, calcein, SYBR Green, eriochrome black T [22] [29] |
| Reverse Transcriptase | cDNA synthesis from RNA templates | Integrated in Bst 3.0 or separate enzyme [29] |
| Reaction Buffer | Optimal conditions for amplification | Typically includes MgSO₄, betaine, dNTPs [33] |
| Positive Controls | Assay validation | Synthetic RNA, inactivated virus [27] |
Applications in Viral Diagnostics
The implementation of LAMP technology offers particular advantages in several diagnostic scenarios:
Point-of-Care Testing
The simplicity and portability of LAMP systems enable reliable molecular testing outside traditional laboratory settings. Researchers have demonstrated complete smartphone-based systems that can detect SARS-CoV-2 in clinical samples with results in 30 minutes, showing 100% agreement with RT-PCR controls [31]. Such systems are particularly valuable for rapid screening in clinics, airports, and remote locations where access to centralized laboratory facilities is limited.
High-Throughput Screening
The minimal hands-on time and rapid results make LAMP suitable for large-scale screening programs. The colorimetric detection format allows for visual assessment without instrumentation, further enhancing its suitability for mass testing scenarios [33]. During the COVID-19 pandemic, several laboratories implemented LAMP-based screening programs to complement their RT-qPCR capabilities, significantly reducing turnaround times for results.
Resource-Limited Settings
The reduced equipment requirements and lower cost per test (approximately €1.7 per sample according to one study [33]) make LAMP particularly suitable for regions with limited laboratory infrastructure. The ability to use dried reagents that remain stable at room temperature for extended periods further enhances its utility in settings without reliable cold chain infrastructure [33].
LAMP technology represents a significant advancement in molecular diagnostics, offering substantial advantages over RT-qPCR in terms of speed, simplicity, and equipment requirements. While RT-qPCR remains the gold standard for quantitative viral load assessment, LAMP provides a robust, rapid, and accessible alternative for qualitative detection, particularly in point-of-care and resource-limited settings. The continued refinement of LAMP protocols, including improved primer design, polymerase engineering, and detection methodologies, will further expand its applications in viral diagnostics. For researchers and drug development professionals, LAMP offers a valuable tool for rapid screening and diagnosis, potentially transforming approaches to outbreak management and infectious disease control.
Loop-mediated isothermal amplification (LAMP) has emerged as a transformative molecular technique for rapid viral diagnostics, particularly valuable in point-of-care (POC) and resource-limited settings. This method utilizes a strand-displacing DNA polymerase and 4-6 specially designed primers that recognize 6-8 distinct regions of the target sequence, enabling highly specific amplification under isothermal conditions (60-65°C) without requiring thermal cycling equipment [22] [34]. The technique has gained significant traction for diagnosing diverse viruses, as demonstrated by its crucial role during the COVID-19 pandemic and recent applications in detecting respiratory viruses, herpesviruses, and other significant pathogens [34].
Compared to quantitative PCR (qPCR), the gold standard in molecular diagnostics, LAMP offers distinct advantages including rapid results (typically 30-60 minutes), minimal equipment requirements, and compatibility with various detection methods ranging from simple colorimetric changes to sophisticated multiplexed platforms [22] [34]. Recent advancements have expanded LAMP's capabilities to include various adaptations such as DARQ-LAMP, QUASR, FLOS-LAMP, displacement probes, and molecular beacons, enabling multiplex detection of multiple targets in a single reaction [34]. This article provides a comprehensive overview of LAMP-based applications in viral pathogen detection, structured protocols for implementation, and emerging trends shaping the future of rapid viral diagnostics.
Current Applications in Viral Pathogen Detection
Respiratory Virus Detection
Respiratory pathogens represent a significant global health burden, with influenza A virus (IAV) and respiratory syncytial virus (RSV) alone causing millions of severe cases annually [35]. Recent research has focused on developing multiplex LAMP assays to address the diagnostic challenges presented by these pathogens, which often cause similar clinical symptoms but require different treatment approaches.
A notable advancement is the development of a dual LAMP-Lateral Flow Device (LFD) assay for simultaneous detection of H1N1 influenza virus and RSV. This method employs a dual-labeled probe system (H1N1: digoxigenin/biotin; RSV: 6-carboxyfluorescein/biotin) combined with a two-color latex microsphere signal system that enables intuitive visual interpretation of multiple detection results. The entire detection process is completed within 40 minutes at a constant temperature of 63°C, demonstrating a limit of detection (LOD) of 7.78 × 10³ copies/mL for H1N1 IAV and 1.29 × 10² copies/mL for RSV [35].
For human adenoviruses (HAdV), particularly types 3 and 7 which cause severe pediatric respiratory infections, a multi-platform LAMP system has been successfully validated. This system incorporates three detection modalities: calcein, immunochromatography (IC), and fluorescent probe methods. The calcein and IC methods achieved an LOD of 2.5 copies/reaction, while the fluorescent probe method demonstrated superior sensitivity with an LOD of 1 copy/reaction and a median Ct value of 7.3, 72.8% lower than that of qPCR [36].
The VirChip platform represents another significant innovation, enabling multiplexed detection of SARS-CoV-2, influenza A, influenza B, and RSV (A/B) with an LOD of 100 RNA copies per reaction. This valve-free, autonomously loading microfluidic platform facilitates rapid, inexpensive, and multiplexed detection, allowing pathogen screening by primary care providers not only in hospitals but also in resource-limited areas [37].
Detection of Other Significant Viral Pathogens
Beyond respiratory viruses, LAMP has been successfully applied to detect various other clinically significant viruses. For the detection of monkeypox virus (MPXV), which the World Health Organization has declared a global health emergency, researchers have developed LAMP assays capable of distinguishing between the two major clades: Congo Basin (Clade-I) and West African (Clade-II). These assays utilize both fluorescence and visible colorimetric readouts, with sensitivities of 10³ and 10⁷ copies, respectively, providing essential tools for precise diagnosis and effective control of Mpox [38].
In the realm of avian influenza surveillance, a novel paper-based LAMP test has been developed for diagnosing the H5 subtype of avian influenza virus (AIV). This inexpensive, user-friendly point-of-need diagnostic tool demonstrates a detection limit of 500 copies per reaction (25 copies/μL) and requires only a water bath for incubation with visual detection of results without special equipment [39].
Similarly, for syphilis detection, a paper-based LAMP assay targeting Treponema pallidum DNA has been developed with a detection limit of 6.4 × 10⁻⁴ ng/μL. Clinical evaluation using 52 suspected syphilis cases and 25 healthy volunteers demonstrated a sensitivity of 96.15% and specificity of 100%, highlighting its potential as a portable, cost-effective lab-on-a-chip diagnostic solution [40].
Table 1: Performance Metrics of Recent LAMP Assays for Viral Pathogen Detection
| Target Pathogen | Detection Method | Limit of Detection (LOD) | Time to Result | Reference |
|---|---|---|---|---|
| H1N1 Influenza A Virus & RSV | Dual LAMP-LFD | H1N1: 7.78 × 10³ copies/mLRSV: 1.29 × 10² copies/mL | 40 minutes | [35] |
| HAdV-3 & HAdV-7 | Multi-platform (Calcein/IC) | 2.5 copies/reaction | ≤20 minutes | [36] |
| HAdV-3 & HAdV-7 | Multi-platform (Fluorescent Probe) | 1 copy/reaction | ≤20 minutes | [36] |
| SARS-CoV-2, Influenza A/B, RSV | VirChip Microfluidic | 100 RNA copies/reaction | Not specified | [37] |
| MPXV (Clade I) | Fluorescence LAMP | 10³ copies | Not specified | [38] |
| MPXV (Clade I) | Colorimetric LAMP | 10⁷ copies | Not specified | [38] |
| H5 Avian Influenza | Paper-based LAMP | 500 copies/reaction | Not specified | [39] |
| Treponema pallidum | Paper-based LAMP | 6.4 × 10⁻⁴ ng/μL | Not specified | [40] |
Experimental Protocols
Dual LAMP-LFD Assay for Simultaneous Detection of H1N1 and RSV
Primer and Probe Design
- Target Selection: Identify highly conserved regions using GenBank nucleic acid sequence database from NCBI. For H1N1, target the hemagglutinin (HA) gene (GenBank: NC007366.1); for RSV, target the fusion (F) protein gene (GenBank: NC001803.1) [35].
- Primer Design: Utilize Primer Explorer V5 to design LAMP primers: two external primers (F3 and B3), two internal primers (FIP and BIP), and loop primers (LF and LB). Employ NUPACK software to predict primer specificity and minimize dimer formation [35].
- Probe Design: Design specific probes for conserved sequences. Modify the 5' of FIPs with biotin, and label specific probes with digoxigenin for H1N1 and 6-carboxyfluorescein for RSV [35].
Reaction Setup
Reaction Composition:
- Bst 2.0 WarmStart DNA Polymerase
- WarmStart RTx Reverse Transcriptase for RNA viruses
- 10× Isothermal Amplification Buffer
- MgSO₄
- dNTPs
- Primers and dual-labeled probes
- Heat-labile UDG enzyme to prevent contamination [35]
Amplification Conditions:
- Temperature: 63°C constant
- Time: 40 minutes
- Platform: Dry block heater or water bath [35]
Detection via Lateral Flow Device
- Assembly: Apply amplified products to the sample pad of the LFD.
- Principle: Biotin-labeled amplicons bind to streptavidin-coated latex microspheres, then migrate to capture lines with specific antibodies (anti-digoxigenin for H1N1, anti-FITC for RSV).
- Interpretation: Visual readout of colored test lines within 5-10 minutes [35].
Multi-Platform LAMP Detection System for HAdV
Primer Design for Hexon Gene Targets
- Sequence Alignment: Conduct multiple sequence alignments of HAdV-3 and HAdV-7 whole-genome sequences from NCBI database using SnapGene software to identify conserved regions of the Hexon gene [36].
- Primer Design: Use PrimerExplorer V5 online platform to design LAMP primer sets including outer primers (F3/B3), inner primers (FIP/BIP), and loop primers (LF/LB) [36].
- Platform-Specific Modifications:
- IC Method: Attach TAMRA fluorescent group to 5' end of FIP primer (FIP-M) and biotin to 5' end of LF primer (LF-M).
- Fluorescent Probe Method: Develop dual-labelled probe (P) with HEX fluorescent group at 5' end and BHQ1 quenching group at 3' end [36].
Reaction Optimization
Master Mix Preparation:
- 2× RT-LAMP Premix 2.0 HS (Probe)
- dNTPs (10 mmol/L)
- Primers with platform-specific modifications
- Betaine and trehalose for stabilization [36]
Amplification Conditions:
- Temperature: 65°C constant
- Time: ≤20 minutes
- Real-time monitoring for fluorescent probe method [36]
Detection Modalities
- Calcein Method: Include calcein in reaction mix; positive amplification shows green fluorescence under UV light or color change from orange to green visible to naked eye.
- Immunochromatography (IC): Use biotin-TAMRA dual-labeled test strips; positive results show visible test lines.
- Fluorescent Probe Method: Monitor real-time fluorescence signals using compatible instrumentation [36].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of LAMP assays requires carefully selected reagents and materials optimized for isothermal amplification. The following table summarizes key components and their functions in LAMP-based viral detection assays.
Table 2: Essential Research Reagents for LAMP-Based Viral Detection
| Reagent/Material | Function | Examples/Specifications |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing enzyme for isothermal amplification | Lyo-ready Bst DNA Polymerase, Bst 2.0 WarmStart, Bst 3.0 [41] |
| Reverse Transcriptase | cDNA synthesis for RNA viruses | WarmStart RTx Reverse Transcriptase, SuperScript IV RT [35] [41] |
| Isothermal Amplification Buffer | Optimal reaction conditions for Bst polymerase | 10× Isothermal Amplification Buffer with MgSO₄ [35] |
| Primer Sets | Target-specific amplification | 4-6 primers per target (F3, B3, FIP, BIP, LF, LB) [22] |
| Detection Probes | Specific signal generation for multiplex detection | Dual-labeled probes (digoxigenin/biotin, FITC/biotin) [35] |
| Lateral Flow Strips | Visual readout of multiplex results | Nitrocellulose membranes with specific capture antibodies [35] |
| Colorimetric Dyes | Visual detection of amplification | Phenol red, hydroxynaphthol blue, calcein [42] [40] |
| Nucleic Acid Extracts | Template material | Purified RNA/DNA or crude samples (heat-inactivated swabs) [37] |
| RNase Inhibitors | Prevent RNA degradation in RT-LAMP | RNaseOUT Recombinant Ribonuclease Inhibitor [41] |
| Stabilizers | Long-term reagent stability | Betaine, trehalose [36] |
Workflow and Signaling Pathways
The following diagrams illustrate key experimental workflows and detection mechanisms in LAMP-based viral detection assays.
LAMP Amplification and Detection Workflow
Dual LAMP-LFD Detection Mechanism
Multi-Platform LAMP Detection System
LAMP technology has evolved into a sophisticated diagnostic platform that balances analytical performance with practical utility for viral pathogen detection. The applications outlined in this article demonstrate how LAMP-based assays deliver rapid, sensitive, and specific detection of clinically relevant viruses across diverse healthcare settings. The dual LAMP-LFD assay for respiratory viruses, multi-platform HAdV detection system, and innovative microfluidic platforms like VirChip represent significant advancements in point-of-care molecular diagnostics.
Future directions in LAMP-based viral detection will likely focus on enhancing multiplexing capabilities, integrating sample preparation steps, developing quantitative readouts, and creating connected digital platforms for result interpretation and data management. As these technologies mature, LAMP-based assays are poised to play an increasingly vital role in global viral pathogen surveillance, outbreak response, and clinical management of infectious diseases.
Advanced LAMP Protocols: Implementation Across Diverse Viral Pathogens and Detection Formats
The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has underscored the critical need for rapid, accurate, and accessible diagnostic tools to facilitate timely public health interventions [43]. While reverse transcription quantitative polymerase chain reaction (RT-qPCR) remains the gold standard for SARS-CoV-2 detection, its requirement for sophisticated laboratory infrastructure, skilled personnel, and lengthy processing times has motivated the development of alternative diagnostic platforms [43] [44]. Reverse transcription loop-mediated isothermal amplification (RT-LAMP) has emerged as a powerful technique that addresses several limitations of RT-qPCR, offering rapid detection (often under 45 minutes), operational simplicity, and minimal equipment requirements while maintaining high sensitivity and specificity [43] [45] [46].
This application note provides detailed protocols and experimental data for detecting SARS-CoV-2 through RT-LAMP assays targeting three essential genomic regions: ORF1ab, nucleocapsid (N) gene, and envelope (E) gene. The multiplex targeting approach enhances detection reliability by providing redundant confirmation and mitigating the impact of viral mutations [46]. We present optimized reaction conditions, performance validation against RT-qPCR, and implementation guidelines suitable for both well-equipped laboratories and resource-limited settings.
Target Selection and Rationale
Strategic selection of target genes is paramount for developing robust SARS-CoV-2 detection assays. Multiplex targeting provides complementary verification, improving detection confidence and protecting against diagnostic escape due to viral mutations.
Table 1: SARS-CoV-2 Genomic Targets for RT-LAMP Detection
| Target Gene | Function | Advantages for Detection | Conservation | References |
|---|---|---|---|---|
| ORF1ab | Encodes replicase polyprotein essential for viral replication | Highly specific to SARS-CoV-2; present in high copies during replication | Highly conserved across variants | [47] [46] |
| N Gene | Encodes nucleocapsid protein packaging viral RNA | Highly expressed during infection; abundant transcript | Moderate conservation with stable regions | [45] [48] [44] |
| E Gene | Encodes envelope protein involved in virion assembly | Essential gene with limited mutation tolerance | Highly conserved across coronaviruses | [43] [44] |
The ORF1ab region, which constitutes approximately two-thirds of the viral genome, contains non-structural proteins involved in replication and exhibits high sequence specificity to SARS-CoV-2 [46]. The N gene is abundantly expressed during infection, making it an ideal target for sensitive detection [45]. The E gene shares conservation across coronaviruses but contains regions unique to SARS-CoV-2, providing a balance between broad detection capability and specificity [43].
Multiplexed detection of these targets significantly enhances diagnostic accuracy. A study evaluating a triplex RT-LAMP assay demonstrated 100% sensitivity and 98.6% specificity compared to RT-qPCR, highlighting the robustness of simultaneous multi-gene detection [46].
Performance Comparison of Detection Methods
Understanding the relative performance characteristics of SARS-CoV-2 detection methodologies enables appropriate test selection based on specific application requirements.
Table 2: Comparative Performance of SARS-CoV-2 Detection Methods
| Method | Sensitivity | Specificity | Time to Result | Cost | Equipment Needs | LoD |
|---|---|---|---|---|---|---|
| RT-qPCR | ~100% [43] | ~100% [43] | 1.5-4 hours [49] | High [43] | Real-time thermal cycler, RNA extraction equipment [43] | 15 copies/reaction [43] |
| RT-LAMP | 84.13-100% [43] [45] [46] | 86.67-100% [43] [45] [44] | 30-45 minutes [43] [46] | Low to moderate [45] | Heating block or water bath, minimal equipment [44] | 0.65-3 copies/μL [46] |
| Antigen Test | 82.46% [45] | 100% [45] | 15-30 minutes [43] | Low [50] | None for visual read tests [43] | 2-3 times higher than RT-qPCR [43] |
RT-LAMP demonstrates particularly strong performance for samples with high viral loads (Ct values <30), with sensitivity reaching 98-100% [43]. One study reported 100% sensitivity for samples with Ct values ≤30, and 84.13% overall sensitivity compared to RT-qPCR [43]. The limit of detection (LoD) for optimized RT-LAMP assays ranges from 0.65 to 3 copies/μL, comparable to RT-qPCR [46].
Modeling studies indicate that testing frequency and turnaround time are more critical than ultimate sensitivity for epidemic control, positioning RT-LAMP as a highly effective tool due to its rapid results and minimal infrastructure requirements [50].
Experimental Protocols
Primer Design and Optimization
Careful primer design is fundamental to successful RT-LAMP assay development. Each primer set should recognize six distinct regions within the target sequence.
ORF1ab Target Primers:
- Follow the design principles established by [46] with modifications for compatibility with multiplex detection
- Target conserved regions within the RNA-dependent RNA polymerase (RdRp) domain
- Validate specificity against circulating variant sequences
N Gene Target Primers:
- Utilize established designs from [44] with modifications for enhanced stability
- Target the structured RNA-binding domain with high conservation
- Optimize concentrations to minimize primer-dimer formation
E Gene Target Primers:
- Implement designs from [44] with adjustments for multiplex compatibility
- Focus on membrane-association domains with sequence uniqueness to SARS-CoV-2
- Verify absence of cross-reactivity with human coronaviruses
Primer mixing should maintain the following concentration ratios in the 10X primer master mix: 16 μM each of FIP and BIP, 2 μM each of F3 and B3, and 4 μM each of LF and LB [43]. Lyophilized primer stocks enhance stability and facilitate distribution to resource-limited settings [46].
RNA Extraction and Sample Preparation
Conventional RNA Extraction:
- Use 100-200 μL of nasopharyngeal swab samples in viral transport medium [44]
- Extract RNA using commercial kits (e.g., GenElute Total RNA Purification kit, QIAamp DSP Virus Kit) following manufacturer protocols [46] [44]
- Elute in 50 μL nuclease-free water and store at -80°C if not used immediately [46]
Extraction-Free Direct Detection:
- Vortex nasopharyngeal swabs in universal transport medium for 15 seconds [44]
- Add proteinase K at 1-2.5 mg/mL concentration and incubate at 55°C for 15-30 minutes [44]
- Heat-inactivate at 95°C for 5-10 minutes to release viral RNA [44]
- Centrifuge briefly and use 5-15 μL of supernatant directly in RT-LAMP reactions [44]
The direct detection method achieves 83.61% sensitivity and 86.67% specificity, providing a viable option when RNA extraction is not feasible [44].
RT-LAMP Reaction Setup
Reagent Composition (20 μL Reaction):
- 12.5 μL WarmStart 2X LAMP Master Mix (includes Bst DNA polymerase) [43]
- 2.5 μL 10X primer mix (containing all three target primer sets) [43]
- 0.5 μL 50X fluorescence dye (for real-time detection) OR phenol red (for colorimetric detection) [43] [44]
- 4.5 μL nuclease-free water [43]
- 5 μL RNA template or direct sample preparation [43]
Reaction Enhancement Additives:
- Supplement with 0.32 U/μL additional Bst 2.0 polymerase to increase reaction speed [49]
- Include 10-25 mM betaine to improve amplification efficiency [44]
- Add 1-2 mM guanidine hydrochloride to enhance specificity and reduce stochastic effects [49]
Thermal Cycling Conditions:
- Incubate at 63°C for 30-45 minutes in a heating block, water bath, or real-time thermal cycler [43]
- For real-time detection, monitor fluorescence every minute for 45 cycles [43]
- Include no-template controls and positive controls in each run [43]
Detection and Analysis
Colorimetric Detection:
- Visual observation of color change from pink to yellow due to pH shift [46] [44]
- Use phenol red indicator in the reaction mix (0.5 μL of 50X) [44]
- Provides simple yes/no results without instrumentation [46]
Fluorescence Detection:
- Monitor real-time amplification using intercalating dyes (GelGreen, SYBR Green) [43] [48]
- Use FAM channel (450-490 nm excitation, 510-530 nm detection) with real-time PCR instruments [43]
- Enables quantification and reaction monitoring [43]
Melting Curve Analysis:
- Program thermal cycler for dissociation curve from 63°C to 95°C post-amplification [43]
- Verify specific amplification by characteristic peak temperature [43]
- Differentiate specific amplicons from non-specific products [43]
Agarose Gel Electrophoresis:
- Separate RT-LAMP products on 2% agarose gel [44]
- Identify characteristic ladder pattern of LAMP amplicons [44]
- Confirm amplification specificity [44]
Research Reagent Solutions
Table 3: Essential Reagents for SARS-CoV-2 RT-LAMP Detection
| Reagent | Function | Recommended Products | Storage Conditions |
|---|---|---|---|
| Bst DNA Polymerase | Strand displacement amplification | WarmStart LAMP Master Mix (NEB) [43], SuperScript IV RT-LAMP Master Mix [48] | -20°C |
| Primer Sets | Target-specific amplification | Custom synthesized (e.g., Gene Fanavaran [44]) with RPC purification | -20°C (lyophilized stable at RT) |
| Reverse Transcriptase | RNA to cDNA conversion | Included in WarmStart and SuperScript master mixes [43] [48] | -20°C |
| Detection Dyes | Signal generation | GelGreen [48], phenol red [44], SYBR Green | -20°C, protected from light |
| Enhancement Additives | Improved sensitivity/specificity | Guanidine HCl [49], betaine [44] | Room temperature |
| RNA Extraction Kits | Nucleic acid purification | GenElute Total RNA Purification [46], QIAamp DSP Virus Kit [44] | Room temperature |
Advanced Applications and Modifications
CRISPR-Cas12a Coupled Detection
Integration of CRISPR-Cas12a with RT-LAMP enables highly specific detection while reducing false positives:
- Perform RT-LAMP amplification as described previously [49]
- Transfer 2 μL of RT-LAMP product to Cas12a reaction mixture [49]
- Incubate at 37°C for 20 minutes with target-specific crRNAs [49]
- Detect collateral cleavage of fluorescent reporter via fluorescence [49]
This approach achieves 100% specificity and 93% sensitivity with LoD of 3 copies/μL [49].
Digital Droplet RT-LAMP (ddRT-LAMP)
Partitioning reactions into droplets enables absolute quantification:
- Generate 105 μm diameter droplets using flow-focusing microfluidics [48]
- Stabilize with fluorinated oil containing 3% w/w fluorinated surfactant [48]
- Incubate at 63°C for 30 minutes [48]
- Image droplets and count positive fractions using Poisson statistics [48]
ddRT-LAMP achieves detection and quantification limits of 10² copies/μL [48].
Troubleshooting and Optimization
Low Sensitivity:
- Increase input volume of RNA template (up to 5 μL in 20 μL reaction) [44]
- Supplement with additional Bst polymerase (0.32 U/μL) [49]
- Include guanidine hydrochloride (1-2 mM) to enhance efficiency [49]
Non-Specific Amplification:
- Redesign primers to avoid secondary structures and dimers [44]
- Optimize magnesium concentration (2-8 mM range) [44]
- Implement touchdown thermal protocols (65°C for 5 min, then 63°C) [44]
Inconsistent Colorimetric Results:
- Prepare fresh master mixes and avoid repeated freeze-thaw cycles [46]
- Use lyophilized reagents for improved stability [46]
- Include internal amplification controls [49]
RT-LAMP represents a robust, rapid, and cost-effective alternative to RT-qPCR for SARS-CoV-2 detection, particularly valuable in resource-limited settings and for point-of-care applications. The multiplex targeting of ORF1ab, N, and E genes provides enhanced detection reliability and coverage against emerging variants. With sensitivities reaching 84.13-100% and specificities of 86.67-100% compared to RT-qPCR, along with the capacity to detect viral RNA with CT values up to 35, RT-LAMP assays offer a compelling diagnostic solution [43] [45] [44]. The protocols outlined in this application note provide researchers with comprehensive methodologies for implementing these assays in diverse laboratory settings.
The resurgence of monkeypox virus (MPXV) and the declaration of a global public health emergency have underscored critical gaps in diagnostic capabilities, particularly the need for rapid and accessible tools that can differentiate between the major viral clades [51] [38]. MPXV is phylogenetically divided into Clade I (historically Congo Basin) and Clade II (West African), with subclades including IIa, IIb, and the newly identified Ib [51]. Clade I is associated with higher virulence and case fatality rates (up to 10%), while Clade II, responsible for the 2022-2024 global outbreaks, typically presents with milder disease but demonstrates heightened transmissibility [52] [51]. This clade-specific variation in pathogenicity and transmission dynamics makes differential detection not just a technical exercise but a crucial component of effective public health response and clinical prognosis [38].
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful alternative to real-time PCR (qPCR), offering rapid amplification at constant temperatures without requiring sophisticated thermal cycling equipment [53] [38]. This technology is particularly suited for low-resource settings and point-of-care applications, providing results with high sensitivity and specificity through fluorescent or colorimetric readouts [54] [55]. Within the context of a broader thesis on LAMP assays for rapid viral diagnostics, this application note details standardized protocols for the multiplexed detection and discrimination of MPXV Clades I and II, enabling researchers to accurately characterize circulating strains with resources appropriate for both laboratory and field settings.
Technical Performance Data
The following tables consolidate performance metrics from recently developed LAMP and PCR assays for MPXV clade discrimination, providing a comparative overview of analytical sensitivities, specificities, and key operational characteristics.
Table 1: Performance Comparison of MPXV Clade-Specific LAMP Assays
| Assay Description | Target Gene | Clade Specificity | Analytical Sensitivity (copies/µL) | Time to Result | Readout Method |
|---|---|---|---|---|---|
| LAMP-1 Assay [38] | D13L, D14L, D15L | Clade I | 10³ (Fluorescence), 10⁷ (Colorimetric) | <60 min | Fluorescent, Colorimetric |
| LAMP-2 Assay [38] | H5R, C7L, C8L | Clade II | 10³ | <60 min | Fluorescent (DARQ probe) |
| Pan-OPV LAMP [54] | A4L, N1R | All Old World Orthopoxviruses | 6.25 | ~30 min | Colorimetric |
| MPXV-Specific LAMP [53] | A27L, F3L | All MPXV (Pan-MPXV) | 20 | 16-55 min | Colorimetric, Turbidity |
Table 2: Performance of Reference PCR Assays for MPXV Detection and Clade Discrimination
| Assay Description | Targets | Purpose | Analytical Sensitivity | Clinical Sensitivity | Clinical Specificity |
|---|---|---|---|---|---|
| MpoxEG4-plex rPCR [52] | Essential OPXV genes | Quadruplex detection: OPXV, MPXV, Clade I, Clade II | ≤9 copies/reaction | >96% | >96% |
| Clade Ib ddPCR [56] | Clade Ib-specific | Wastewater surveillance | ≈1,000 copies/g (dry weight) | N/A | N/A |
| Multiplex rPCR [57] | N3R, B18Rplus (MPXV); E9L, D6R (OPXV) | Simultaneous MPXV & Orthopoxvirus detection | copy/reaction | N/A | N/A |
Experimental Protocols
LAMP Assay for Clade I and Clade II Discrimination
This protocol is adapted from the development of LAMP-1 (Clade I) and LAMP-2 (Clade II) assays, which utilize distinct primer sets and probe technologies for specific clade identification [38].
Primer and Probe Design
- Clade I (LAMP-1) Targets: Design primers targeting conserved and clade-specific regions within the D13L, D14L, and D15L genes of Clade I MPXV genomes.
- Clade II (LAMP-2) Targets: Design primers targeting conserved and clade-specific regions within the H5R, C7L, and C8L genes of Clade II MPXV genomes.
- Probe Selection for LAMP-2: Incorporate a Novel R-Duplex DARQ probe for specific detection of Clade II amplicons via fluorescence, enhancing specificity and reducing false positives [38].
- Bioinformatic Validation: Confirm primer specificity by performing BLAST analysis against representative sequences from both clades and other orthopoxviruses to ensure no cross-reactivity.
Reaction Setup
- LAMP Master Mix (25 µL total volume):
- 1× Isothermal Amplification Buffer (e.g., WarmStart LAMP Buffer)
- 6-8 mM MgSO₄ (optimal concentration requires empirical testing)
- 1.4 mM dNTP mix
- 0.8 M Betaine (enhances strand displacement)
- 0.2 µM F3 and B3 primers (outer primers)
- 1.6 µM FIP and BIP primers (inner primers)
- 0.4 µM LF and LB primers (loop primers, if designed)
- 8 U Bst 2.0 WarmStart DNA Polymerase
- For colorimetric readout: Add 1× Phenol Red or 120 µM Hydroxynaphthol Blue
- For fluorescent LAMP-2: Add 0.2 µM R-Duplex DARQ probe
- 5 µL DNA template
- Nuclease-free water to 25 µL
Amplification Conditions
- Temperature: Incubate reaction at 63-65°C for 45-60 minutes [53] [54].
- Enzyme Activation: A preliminary 30-second incubation at 95°C may be used for WarmStart enzymes, followed by immediate transfer to isothermal temperature.
- Reaction Termination: Heat inactivation at 80°C for 5 minutes.
Result Interpretation
- Colorimetric Readout: Positive reaction changes from pink to yellow. Include a negative control (water) that remains pink and a positive control (synthetic DNA) that turns yellow [54].
- Fluorescent Readout: Monitor real-time fluorescence in FAM or SYBR Green channels. A positive reaction shows a characteristic sigmoidal amplification curve crossing the threshold within 30-40 minutes [38].
- Specificity Confirmation: Verify Clade II amplification with the DARQ probe fluorescence channel distinct from the intercalating dye signal.
Direct Detection from Swab Samples Without Nucleic Acid Extraction
This extraction-free protocol enables rapid testing from clinical specimens, significantly reducing processing time and resource requirements [54].
Sample Processing
- Sample Collection: Collect lesion swabs in universal or viral transport media.
- Sample Inactivation: Mix 200 µL of swab eluate with 200 µL of AL lysis buffer (Qiagen) and 20 µL of protease. Incubate at 56°C for 15 minutes [52].
- Direct Template Preparation: Centrifuge the inactivated sample at 12,000 × g for 2 minutes. Use 5-10 µL of the supernatant directly as template in the LAMP reaction.
Optimization Notes
- Inhibition Management: If inhibition is observed, dilute the supernatant 1:2 to 1:5 in nuclease-free water before adding to the LAMP reaction.
- Sensitivity Validation: Compare with extracted samples to establish any potential sensitivity reduction with direct detection.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials for MPXV LAMP Assays
| Reagent/Material | Function | Specifications/Alternatives |
|---|---|---|
| Bst 2.0 WarmStart DNA Polymerase | Isothermal strand displacement amplification | Select high-activity formulations for rapid amplification |
| LAMP Primer Sets (F3/B3, FIP/BIP, LF/LB) | Specific target recognition and amplification | HPLC-purified; validate specificity in silico and empirically |
| Phenol Red or HNB Dye | Colorimetric pH indicator for visual readout | Pre-optimized concentration in reaction buffer |
| R-Duplex DARQ Probes | Fluorescent detection for specific clade identification | Quencher-fluorophore systems compatible with LAMP chemistry |
| Synthetic MPXV DNA Controls | Positive controls for Clade I and Clade II | G-block genes encompassing target regions with clade-specific SNPs |
| Universal Transport Media | Clinical sample preservation and viral inactivation | Compatible with direct detection protocols without extraction |
| GuHCl (Guanidine Hydrochloride) | Reaction additive to improve amplification speed | 40 mM final concentration; optimize for each primer set |
Discussion
The developed protocols demonstrate that LAMP-based assays provide a robust platform for discriminating between MPXV Clade I and Clade II with sensitivities approaching or matching those of reference PCR methods [52] [38]. The strategic targeting of clade-specific genes in the central conserved region of the MPXV genome ensures reliable detection despite ongoing viral evolution, which has compromised some assays targeting terminal regions [52]. The integration of extraction-free protocols further enhances the utility of these assays in resource-limited settings, potentially reducing turnaround time by several hours.
Critical implementation considerations include the trade-off between simplicity and multiplexing capability. While the described LAMP assays can run in parallel for clade discrimination, the recently developed MpoxEG4-plex rPCR demonstrates the potential for simultaneous detection of orthopoxvirus, MPXV species, and clade assignment in a single reaction [52]. However, this requires sophisticated instrumentation less accessible in point-of-care contexts. For surveillance applications, the detection of MPXV Clade Ib in wastewater using specific PCR assays highlights another application where these discriminatory tests provide public health utility [56].
Future development directions should focus on truly multiplexed LAMP formats that can distinguish clades in a single tube, potentially through multicolor fluorescence detection or microfluidic partitioning. Additionally, the emergence of new subclades necessitates continuous monitoring of primer complementarity and inclusivity. Integration of these assays with portable reading devices and smartphone-based result interpretation could further expand their deployment in diverse healthcare settings, strengthening global mpox surveillance and response capabilities.
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful nucleic acid amplification technique that is revolutionizing rapid viral diagnostics. As a core component of our broader thesis on LAMP assays for rapid viral detection, this document details the development and application of flexible detection platforms capable of meeting diverse diagnostic needs. Unlike conventional PCR that requires thermal cycling, LAMP amplifies nucleic acids at a constant temperature (60-65°C) using a DNA polymerase with high strand displacement activity and 4-6 primers recognizing 6-8 distinct regions of the target DNA [22] [58]. This isothermal nature eliminates the need for sophisticated instrumentation, making LAMP ideally suited for both laboratory and point-of-care settings [59].
The versatility of LAMP extends to its detection methods, which can be tailored to specific application requirements. We have developed and optimized three principal platforms—colorimetric, fluorescent, and real-time systems—that form a hierarchical detection network for various diagnostic scenarios. These platforms enable "preliminary screening-quantitative" detection capabilities, from simple visual assessment to precise quantification [36]. The following sections provide detailed application notes and experimental protocols for implementing these flexible detection formats in viral diagnostics.
LAMP Principles and Advantages
Fundamental Mechanism
The LAMP reaction employs a sophisticated primer system consisting of inner primers (FIP, BIP), outer primers (F3, B3), and optional loop primers (LF, LB) that collectively recognize up to eight distinct regions on the target DNA [22] [2]. This multi-primer approach contributes to exceptionally high specificity. The reaction proceeds through a complex mechanism involving initial template formation, cycling amplification, and elongation stages, ultimately generating characteristic cauliflower-like DNA structures with multiple loops [22] [2].
A key enabler of LAMP technology is the Bst DNA polymerase derived from Geobacillus stearothermophilus, which exhibits strong strand displacement activity at constant temperatures of 60-65°C [22] [2]. This enzyme property eliminates the need for thermal denaturation steps required in PCR. Recent engineering efforts have further enhanced Bst polymerase characteristics, with newer versions such as Bst-XT WarmStart, Bst 2.0, and Bst 3.0 offering improved speed, sensitivity, and resistance to inhibitors [58] [41].
Advantages Over PCR-Based Methods
LAMP offers several compelling advantages for viral diagnostics compared to traditional PCR methods. The technique can produce up to 10⁹ copies of amplified DNA in less than an hour, exceeding the amplification efficiency of conventional PCR [22]. The reaction's robustness provides greater tolerance to inhibitors present in biological samples, potentially enabling simplified nucleic acid extraction or even direct sample analysis [41] [28]. Furthermore, LAMP's isothermal nature eliminates the requirement for expensive thermal cyclers, reducing both initial instrumentation costs and operational complexity [59] [2].
Multi-Platform Detection Systems
Platform Comparison and Performance Characteristics
We systematically evaluated three LAMP detection platforms for the identification of human adenovirus types 3 and 7 (HAdV-3 and HAdV-7), demonstrating how this multi-platform approach creates a hierarchical detection network for varied clinical settings [36]. The performance characteristics of each platform are summarized in Table 1.
Table 1: Performance comparison of LAMP detection platforms for HAdV detection
| Detection Platform | Limit of Detection (copies/reaction) | Specificity (%) | Approximate Detection Time | Best Application Setting |
|---|---|---|---|---|
| Colorimetric (Calcein) | 2.5 | 100 | ≤20 minutes | Grassroots clinics, point-of-care |
| Immunochromatography | 2.5 | 100 | ≤20 minutes | Field testing, outbreak settings |
| Fluorescent Probe | 1 | 100 | ≤20 minutes | Central laboratories, quantitative needs |
The exceptional sensitivity of the fluorescent probe method is evidenced by a median Ct value of 7.3, which was 72.8% lower than that of qPCR (median Ct 26.9; p < 0.05) in clinical validation studies [36]. All three platforms demonstrated 100% specificity with no cross-reactivity against SARS-CoV-2 or other common respiratory pathogens [36].
Colorimetric Platform
Principle and Mechanisms
The colorimetric LAMP platform enables visual detection of amplification through distinct color changes mediated by different mechanisms. The pH-sensitive method utilizes phenol red or similar pH indicators that change color from red to yellow as amplification progresses [25]. This color shift results from the release of hydrogen ions during DNA polymerization, which lowers the pH of the weakly buffered reaction mixture [25] [41].
Metal ion indicators represent another colorimetric approach. Calcein forms a complex with magnesium ions (Mg²⁺) present in the reaction buffer, producing a visible color change under UV light [22] [36]. Hydroxynaphthol blue (HNB) functions similarly, changing from violet to sky blue as magnesium ions are incorporated into magnesium pyrophosphate precipitates during DNA synthesis [22] [59].
Experimental Protocol
Materials Required:
- WarmStart Colorimetric LAMP 2X Master Mix (includes pH-sensitive dye)
- LAMP primers (FIP, BIP, F3, B3, LF, LB)
- Template DNA/RNA
- Nuclease-free water
- Heating block or water bath (65°C)
Procedure:
- Prepare reaction mixture:
- 12.5 μL WarmStart Colorimetric LAMP 2X Master Mix
- 2.5 μL primer mix (final concentration: 1.6 μM FIP/BIP, 0.2 μM F3/B3, 0.8 μM LF/LB)
- 5 μL template DNA/RNA
- Adjust to 25 μL with nuclease-free water
Incubate at 65°C for 20-45 minutes
Visual assessment:
- Positive reaction: Yellow color
- Negative reaction: Pink/Magenta color
- For calcein-based detection: Green fluorescence under UV light indicates positive reaction
Troubleshooting Notes:
- False positives may occur with extended incubation (>45 minutes) due to spurious amplification [25]
- Color interpretation can be standardized using mobile applications for consistent results [28]
- Sample inhibitors may delay color change; include positive and negative controls in each run
Fluorescent Platform
Principle and Mechanisms
The fluorescent LAMP platform employs DNA-intercalating dyes or specialized probes to detect amplification. SYBR Green, EvaGreen, and SYTO-9 are common intercalating dyes that exhibit minimal fluorescence when free in solution but emit strong fluorescence upon binding to double-stranded DNA amplification products [22] [41].
Probe-based detection methods offer enhanced specificity. The Novel R-Duplex DARQ (Detection of Amplification by Release of Quenching) probe system provides highly specific detection capable of distinguishing between closely related viral clades [38]. For immunochromatographic detection, a biotin-TAMRA dual-labeling system enables rapid on-site visualization, where FIP primers are labeled with TAMRA fluorescent group and LF primers with biotin [36].
Experimental Protocol
Materials Required:
- 2X RT-LAMP Premix 2.0 HS
- Fluorescent dye (SYTO-9, EvaGreen) or specific probes
- LAMP primers (appropriately modified for probe-based detection)
- Template DNA/RNA
- Real-time isothermal instrument or endpoint fluorescence reader
Procedure:
- Prepare reaction mixture:
- 12.5 μL 2X RT-LAMP Premix
- 2.5 μL primer mix (concentrations as in colorimetric method)
- 1 μL fluorescent dye (if using intercalating dye) or 0.5-1 μL probe
- 5 μL template
- Adjust to 25 μL with nuclease-free water
For real-time monitoring:
- Incubate at 65°C for 30-60 minutes with continuous fluorescence monitoring
- Set fluorescence acquisition appropriate for dye/probe used
For endpoint detection:
- Incubate at 65°C for 30-45 minutes
- Measure fluorescence using appropriate instrumentation
- Alternatively, visualize under blue light transilluminator for intercalating dyes
Troubleshooting Notes:
- Probe-based assays require careful primer and probe design to avoid interference
- Intercalating dyes may inhibit amplification at high concentrations; optimize concentration
- For multiplex detection, use probes with non-overlapping emission spectra
Real-Time Quantitative Platform
Principle and Mechanisms
Real-time LAMP platforms combine the simplicity of isothermal amplification with quantitative capabilities. These systems employ fluorescent probes or DNA-intercalating dyes to monitor amplification kinetics in real time, similar to quantitative PCR [36] [41]. The process generates amplification curves that allow determination of the time to positivity (Tp), which correlates with the initial template concentration [41].
Advanced probe technologies like the DARQ system enable highly specific real-time monitoring while reducing false positives from non-specific amplification [38]. This approach is particularly valuable for distinguishing between viral clades with high phylogenetic similarity, such as MPXV Clade-I and Clade-II, which have different clinical implications [38].
Experimental Protocol
Materials Required:
- Bst-XT WarmStart DNA Polymerase or equivalent
- dNTP mix
- Reaction buffer (including MgSO₄)
- Fluorescently labeled probes (HEX, FAM, Cy5, etc.)
- LAMP primers
- Template DNA/RNA
- Real-time isothermal instrument (e.g., Axxin T-series)
Procedure:
- Prepare reaction mixture:
- 2.5 μL 10X isothermal amplification buffer
- 1.5 μL MgSO₄ (8 mM final)
- 3.5 μL dNTP mix (1.4 mM each)
- 1 μL Bst-XT WarmStart DNA Polymerase (8 U)
- 2.5 μL primer mix (concentrations as above)
- 0.5-1 μL fluorescent probe (200-500 nM final)
- 5 μL template
- Adjust to 25 μL with nuclease-free water
Reaction conditions:
- Incubate at 65°C for 60 minutes with fluorescence acquisition every 30-60 seconds
- Set appropriate fluorescence channels for probes used
Data analysis:
- Determine Tp (time to positivity) values from amplification curves
- Generate standard curve using known template concentrations for quantification
Troubleshooting Notes:
- High background fluorescence may indicate probe degradation; prepare fresh probes
- Shallow amplification curves suggest suboptimal reaction conditions; optimize Mg²⁺ concentration
- Include no-template controls to identify contamination issues
Research Reagent Solutions
Successful implementation of LAMP detection platforms requires carefully selected reagents optimized for isothermal amplification. Table 2 summarizes key reagent solutions and their specific functions in LAMP assays.
Table 2: Essential research reagents for LAMP assay development
| Reagent Category | Specific Examples | Function and Characteristics |
|---|---|---|
| DNA Polymerases | Bst-XT WarmStart, Bst 2.0, Bst 3.0 | Strand-displacing activity; engineered for speed, sensitivity & inhibitor tolerance [58] [41] |
| Master Mixes | WarmStart Colorimetric LAMP 2X Master Mix, SuperScript IV RT-LAMP Master Mix | Complete optimized mixtures with dyes; include reverse transcriptase for RNA targets [58] [41] |
| Detection Dyes | Phenol red, Calcein, Hydroxynaphthol Blue | Colorimetric indicators for visual detection [22] [25] |
| Fluorescent Dyes | SYTO-9, EvaGreen, SYBR Green | DNA intercalating dyes for fluorescent detection [22] [41] |
| Specialized Probes | DARQ probes, Dual-labeled probes (HEX/BHQ1) | Sequence-specific detection; enhanced specificity; real-time monitoring [36] [38] |
| Primer Design Tools | NEB LAMP Primer Design Tool, PrimerExplorer | Facilitate complex primer design for 6-8 target regions [58] |
Application Workflows
Integrated Diagnostic Pathway
The selection of an appropriate LAMP detection platform depends on the specific diagnostic scenario, including available infrastructure, required throughput, and need for quantification. Figure 2 illustrates a comprehensive workflow integrating all three detection platforms within a coordinated diagnostic pathway.
Platform Selection Guidelines
Colorimetric Platform Applications:
- Point-of-care testing in grassroots healthcare settings
- Rapid screening in resource-limited environments
- Educational settings with minimal equipment availability
- Initial outbreak investigations requiring immediate results
Fluorescent Platform Applications:
- Field testing with portable fluorescence readers
- Medium-throughput testing in regional laboratories
- Detection requiring higher sensitivity than visual methods
- Scenarios benefiting from equipment-based objectivity while maintaining simplicity
Real-Time Quantitative Platform Applications:
- Central laboratories with advanced instrumentation
- Clinical trials requiring viral load quantification
- Genotyping and clade differentiation [38]
- Monitoring treatment efficacy through viral load changes
- Research applications requiring kinetic data
Technical Considerations
Primer Design and Optimization
The success of any LAMP assay fundamentally depends on careful primer design. The process requires selecting 6-8 distinct regions within the target sequence for F3, B3, FIP, BIP, and optional LF/LB primers [22] [2]. Key considerations include:
- Target Selection: Identify conserved regions across viral strains to ensure broad detection capability while avoiding regions with high sequence similarity to non-target organisms [36]
- Software Tools: Utilize specialized LAMP primer design tools such as PrimerExplorer, NEB LAMP Primer Design Tool, or LAMP Designer for initial design [22] [58]
- Validation: Verify primer specificity using BLAST analysis against relevant databases
- Modifications: For probe-based assays, incorporate appropriate modifications (biotin, fluorescent labels) during synthesis [36]
Sensitivity and Specificity Enhancement
Several strategies can enhance LAMP assay performance:
- Loop Primers: Incorporating LF and LB primers can reduce reaction time by up to 50% and improve sensitivity [22] [2]
- Probe Systems: Sequence-specific probes significantly reduce false positives from non-specific amplification [38]
- Enzyme Selection: Newer engineered Bst polymerases offer improved characteristics including faster kinetics and greater resistance to inhibitors [41]
- Additives: Betaine can improve amplification efficiency for GC-rich targets; additional Mg²⁺ may optimize reactions with high DNA concentrations
Multiplexing Capabilities
Multiplex LAMP (M-LAMP) enables simultaneous detection of multiple pathogens or differentiation of viral clades in a single reaction [22] [38]. Implementation strategies include:
- Primer Design: Carefully design primer sets with similar annealing temperatures but distinct target sequences
- Probe-Based Multiplexing: Use differently labeled probes for each target with non-overlapping emission spectra
- Endpoint Differentiation: Employ melting curve analysis after amplification to distinguish products based on Tm differences
The development of flexible colorimetric, fluorescent, and real-time LAMP platforms represents a significant advancement in viral diagnostics, creating a hierarchical detection network suitable for diverse application settings. The protocols and application notes presented herein provide researchers with comprehensive methodologies for implementing these platforms in both laboratory and point-of-care environments.
The colorimetric platform offers simplicity and rapid visual detection ideal for resource-limited settings, while the fluorescent platform provides enhanced sensitivity for field applications. The real-time quantitative platform delivers precise quantification for central laboratories and research applications. Together, these complementary technologies establish a versatile toolkit for addressing current and emerging viral diagnostics challenges, from routine surveillance to outbreak response.
As LAMP technology continues to evolve, future developments will likely focus on further integration with digital platforms, enhanced multiplexing capabilities, and simplified sample preparation methods. These advancements will strengthen the position of LAMP as a cornerstone technology in the molecular diagnostics landscape, particularly for rapid viral detection in diverse healthcare settings.
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful molecular technique for rapid viral diagnostics, offering high sensitivity and specificity without the need for thermal cycling. This application note details novel methodologies in enhanced reverse transcription LAMP (ERT-LAMP) and probe-based detection systems, providing researchers and drug development professionals with advanced tools for pathogen detection. These methodologies address critical limitations of traditional LAMP, including non-specific amplification and inability to perform multiplex detection, while maintaining the technique's inherent advantages for point-of-care applications [60] [61].
The integration of specialized probes and primer design strategies has significantly improved the accuracy and application scope of LAMP assays, making them particularly valuable for rapid diagnosis in both clinical and field settings. This document provides detailed protocols and technical specifications for implementing these advanced LAMP methodologies in viral diagnostics research [36].
Probe-based LAMP methodologies represent a significant advancement over traditional intercalating dye-based detection by providing target-specific verification of amplification products. These systems mitigate false-positive results from non-specific amplification while enabling real-time monitoring and multiplex detection capabilities [60].
Table 1: Comparison of Major Probe-Based LAMP Detection Methodologies
| Methodology | Key Feature | Detection Mechanism | Advantages | Limitations |
|---|---|---|---|---|
| Assimilating Probes [60] | Two partially complementary oligonucleotides | Quencher displacement releases fluorescence | High specificity through dual probe system | More complex probe design required |
| DARQ (Detection of Amplification by Release of Quenching) [60] | Fluorophore-labeled probe complementary to FIP | Strand displacement releases fluorescent probe | Simplified design with single reaction tube | Potential for background signal |
| Q Probe (Quenching Probe) [60] | Fluorophore-labeled cytosine at primer end | Guanine residue in target causes quenching | Inverse signal detection (decrease = positive) | Requires specific sequence context (G) |
| Enzyme-Mediated Probes [60] | Utilizes additional enzymes (endonuclease) | Enzyme cleavage releases signal | Potential for enhanced specificity | Increased cost and reaction complexity |
Advanced Primer Design for Specific Detection
A novel primer modification approach for detecting genetic variations involves strategic alterations at the 3' end of F2 or B2 primers. This design ensures amplification occurs only when the target mutation is present, providing exceptional specificity for discriminating between wild-type and mutated sequences [62].
In this methodology, 2-7 nucleotides at the 3'-end of the F2 primer are designed to match the sequence immediately downstream of the genetic variant. This creates a primer that is specific only to the mutated sequence, preventing amplification of wild-type DNA. The approach has been successfully applied to detect EGFR mutations in non-small-cell lung cancer, demonstrating high specificity and sensitivity even in samples containing mixed wild-type and mutated material [62].
Quantitative Performance Data
Recent validation studies demonstrate the exceptional performance of advanced LAMP systems across various detection platforms. The multi-platform approach enables researchers to select appropriate methodologies based on specific application requirements, from field testing to laboratory quantification [36].
Table 2: Performance Metrics of Multi-Platform LAMP Detection Systems [36]
| Detection Platform | Limit of Detection (LOD) | Specificity | Time to Result | Optimal Application Context |
|---|---|---|---|---|
| Calcein Method | 2.5 copies/reaction | 100% | ≤20 minutes | Field testing, resource-limited settings |
| Immunochromatography (IC) | 2.5 copies/reaction | 100% | ≤20 minutes | Point-of-care testing, rapid screening |
| Fluorescent Probe | 1 copy/reaction | 100% | ≤20 minutes | Laboratory quantification, high-sensitivity needs |
The fluorescent probe method demonstrated superior sensitivity in direct comparisons, with a median Ct value 72.8% lower than that of qPCR (median Ct 7.3 vs. 26.9; p < 0.05), highlighting its exceptional amplification efficiency [36]. Clinical validation across 188 samples showed 100% concordance between fluorescent probe LAMP and qPCR (κ = 1.00; 95% CI: 1.00–1.00) [36].
Detailed Experimental Protocols
Real-Time Fluorescent Probe LAMP Assay
Principle: This protocol utilizes dual-labeled probes with a 5' fluorophore (HEX) and 3' quencher (BHQ1) that hybridize to specific sequences within LAMP amplicons. During amplification, the probe is cleaved or displaced, separating the fluorophore from the quencher and generating a fluorescent signal [36].
Reagent Composition:
- 2× RT-LAMP Premix 2.0 HS (Probe) - 12.5 μL
- Outer primers (F3/B3, 10 μM each) - 0.5 μL each
- Inner primers (FIP/BIP, 40 μM each) - 0.5 μL each
- Loop primers (LF/LB, 20 μM each) - 0.5 μL each
- Dual-labeled probe (P, 10 μM) - 0.5 μL
- Template DNA - 2-5 μL
- Nuclease-free water to 25 μL total volume
Thermal Cycling Protocol:
- Preparation: Mix reagents gently by inversion
- Amplification: Incubate at 65°C for 25-40 minutes
- Detection: Monitor real-time fluorescence in FAM/HEX channels
- Termination: Heat inactivation at 80°C for 5 minutes
Data Interpretation: Positive amplification is indicated by an exponential fluorescence curve crossing the threshold value. The time to positivity (TTP) can be used for quantitative analysis [36] [63].
Multiplex LAMP for Respiratory Pathogen Detection
Principle: This protocol enables simultaneous detection of multiple pathogens in a single reaction by utilizing pathogen-specific primers and probes. The assay targets conserved genomic regions for reliable identification [64].
Sample Processing Workflow:
Pathogen-Specific Target Genes: [64]
- E. coli: phoA gene
- S. aureus: femA gene
- P. aeruginosa: oprL gene
- K. pneumoniae: phoE gene
- S. maltophilia: hrpA gene
- A. baumannii: OXA-51 β-lactamase gene
Performance Characteristics: Clinical validation demonstrated 93.3% sensitivity and 92.0% specificity compared to traditional culture methods, with 93.0% overall accuracy. A weak negative correlation was observed between bacterial load and time to positivity (r = -0.177, p = 0.05) [64].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Advanced LAMP Assay Development
| Reagent/Chemical | Function/Application | Example Use Case |
|---|---|---|
| Bst 2.0/5.0 DNA Polymerase | Strand-displacement polymerase for isothermal amplification | Core enzyme for all LAMP reactions [60] |
| Calcein-Mn²⁺ Complex | Colorimetric/fluorescent detection via metal ion displacement | Visual detection of amplification [36] [65] |
| Hydroxy Naphthol Blue (HNB) | Metal indicator for colorimetric detection | Pre-amplification incorporation for visual readout [61] |
| Dual-Labeled Probes (HEX/BHQ1) | Sequence-specific fluorescence detection | Real-time monitoring in probe-based LAMP [36] |
| Biotin-TAMRA System | Immunochromatographic detection | Lateral flow test strip development [36] |
| RALF Buffer | Rapid DNA extraction and purification | Direct processing of clinical samples [64] |
Troubleshooting and Optimization Guidelines
Primer Design Considerations:
- Utilize PrimerExplorer V5 for initial primer design
- Modify 3' ends of F2/B2 primers for genetic variation detection [62]
- Incorporate TTTT bridges between F1c and F2 sequences
- Verify specificity using NCBI Primer-BLAST
Reducing Non-Specific Amplification:
- Optimize Mg²⁺ concentration (typically 4-8 mM)
- Include 40 mM guanidine hydrochloride to enhance specificity [63]
- Utilize probe-based detection to eliminate false positives from primer dimers
- Implement hot-start enzymes to prevent pre-amplification activity
Multiplexing Challenges:
- Balance primer concentrations to avoid competition
- Minimize primer-primer interactions through careful design
- Validate each target independently before multiplexing
- Consider reagent limitations in complex reaction mixtures [64]
The integration of probe-based detection systems and enhanced primer design strategies has significantly advanced LAMP technology for viral diagnostics. These novel methodologies provide robust, sensitive, and specific detection capabilities suitable for both laboratory and point-of-care applications. The multi-platform approach enables researchers to implement hierarchical testing networks ranging from preliminary screening to quantitative analysis, addressing diverse diagnostic needs across healthcare settings [36].
Further development in multiplexing capabilities and sample processing efficiency will continue to expand the applications of ERT-LAMP and probe-based systems in infectious disease diagnostics, oncology, and environmental testing.
Loop-mediated isothermal amplification (LAMP) has emerged as a transformative technology for molecular diagnostics, particularly for rapid viral detection at the point-of-care. This application note details integrated workflows that seamlessly connect sample processing to result interpretation, enabling laboratories to implement robust testing protocols for respiratory viral pathogens. The protocols outlined herein are designed for researchers and scientists developing diagnostic solutions for pathogens such as SARS-CoV-2, influenza A/B, and respiratory syncytial virus (RSV), with emphasis on workflow integration, sensitivity, and operational simplicity suitable for both clinical laboratories and resource-limited settings.
Performance Comparison of Integrated LAMP Platforms
The table below summarizes the performance characteristics of recently developed LAMP-based diagnostic platforms that integrate sample processing with detection.
Table 1: Performance comparison of integrated LAMP platforms for viral detection
| Platform Name | Target Pathogens | Sample Processing | Amplification Time | Detection Method | Limit of Detection | Reference |
|---|---|---|---|---|---|---|
| VirChip | SARS-CoV-2, Influenza A/B, RSV | Direct crude nasal swab | <60 minutes | Fluorescent detection | 100 RNA copies/reaction | [37] |
| SMART-LAMP | SARS-CoV-2 | Automated RNA purification | Not specified | Fluorescent detection | Comparable to RT-qPCR | [66] |
| LAMP-LFD | H1N1, RSV | Direct processing | 40 minutes | Lateral flow device | H1N1: 7.78×10³ copies/mLRSV: 1.29×10² copies/mL | [35] |
| SPID-LFIA | Bacterial targets (E. coli) | Integrated DNA extraction | 60-75 minutes | Lateral flow immunoassay | 100% sensitivity, 85% specificity | [67] |
Integrated Experimental Workflow
The diagram below illustrates the complete integrated workflow from sample collection to result interpretation, incorporating the key steps validated across multiple LAMP platforms.
Detailed Experimental Protocols
VirChip Platform for Multiplexed Respiratory Virus Detection
The VirChip platform enables valve-free, autonomous loading of microfluidic channels for simultaneous detection of multiple respiratory pathogens [37].
Materials and Reagents
Table 2: Essential research reagents for LAMP-based detection
| Reagent/Component | Function | Example Source/Concentration |
|---|---|---|
| WarmStart LAMP Kit (DNA & RNA) | Isothermal amplification | New England Biolabs (E1700S) |
| WarmStart RTx Reverse Transcriptase | cDNA synthesis for RNA viruses | New England Biolabs (M0380L, 15,000 U/mL) |
| WarmStart Bst 2.0 Polymerase | Strand displacement DNA polymerase | New England Biolabs (M0538S, 8,000 U/mL) |
| LAMP Fluorescent Dye | Real-time detection of amplification | New England Biolabs (B1700) or EvaGreen |
| Primer Mix (F3, B3, FIP, BIP, LF, LB) | Target-specific amplification | Custom designed, 1.6-1.8 µM FIP/BIP |
| Betaine | Stabilizer for amplification efficiency | 0.8 M final concentration |
| dNTP Solution | Nucleotides for DNA synthesis | 1.4 mM final concentration |
| MgSO₄ | Cofactor for polymerase activity | 6-8 mM final concentration |
Microfluidic Device Fabrication
- Master Mold Fabrication: Create a two-layer master mold using a μMLA maskless aligner and standard photolithographic techniques [37].
- PDMS Replication: Cast polydimethylsiloxane (PDMS) replicas using Sylgard 184 elastomer kit (10:1 ratio polymer base to curing agent).
- Device Assembly: Punch 3.0 mm diameter inlets for sample introduction using a biopsy punch.
- Primer Preloading: Preload LAMP primer mixtures specific to target pathogens (SARS-CoV-2, influenza A, influenza B, RSV A/B) into individual microchambers and air-dry.
Sample Processing and Amplification
- Sample Preparation: Crude nasal swab samples can be applied directly to the chip without RNA isolation. For purified RNA, use 5-10 µL input volume.
- Chip Loading: Apply sample to the inlet reservoir. Autonomous degas-driven flow distributes sample to preloaded reaction chambers.
- Amplification Conditions: Incubate at 65°C for 45-60 minutes using a portable heating block or thermal station.
- Real-time Monitoring: Monitor fluorescence accumulation using a portable reader or smartphone-based detection system.
Dual LAMP-LFD for Simultaneous H1N1 and RSV Detection
This protocol enables visual, multiplex detection using lateral flow devices for result interpretation [35].
Primer and Probe Design
- Target Selection: Identify highly conserved regions of H1N1 HA gene (GenBank: NC007366.1) and RSV F protein gene (GenBank: NC001803.1).
- LAMP Primer Design: Design six primers (F3, B3, FIP, BIP, LF, LB) using Primer Explorer V5.
- Probe Modification:
- Modify 5' end of FIP primers with biotin
- Design specific probes labeled with digoxigenin (H1N1) or FITC (RSV)
- Specificity Verification: Use NUPACK software to predict and minimize primer-dimer formation.
LAMP Reaction Setup
Reaction Composition:
- 12.5 µL 2× WarmStart LAMP Master Mix
- 1.5 µL primer mix (all six primers, total concentration 1.6-2.0 µM each)
- 0.5 µL Syto9 (1 µM final) or fluorescent dye
- 5.5 µL nuclease-free water
- 5.0 µL template DNA
- Total volume: 25 µL
Amplification Conditions:
- Incubate at 63°C for 40 minutes
- Use a simple heating block or water bath
- No initial heat denaturation required
Lateral Flow Detection
Device Preparation: Custom LFD with two test lines:
- H1N1: Anti-digoxin antibody line
- RSV: Anti-FITC antibody line
- Control: Anti-biotin antibody line
Sample Application:
- Dilute 5 µL LAMP product in 100 µL running buffer
- Apply to sample pad and allow to migrate for 5-10 minutes
Result Interpretation:
- Positive: Visible test line plus control line
- Negative: Control line only
- Invalid: No control line appearance
Technical Considerations for Workflow Integration
Sample Processing Compatibility
Integrated LAMP workflows must balance simplicity with sensitivity. The VirChip platform demonstrates that direct application of crude nasal swab samples is feasible, eliminating RNA isolation steps [37]. Alternatively, the SMART-LAMP system employs a single-step RNA purification using LyseNtact lysis buffer with centrifugation, providing higher purity template while maintaining operational simplicity [66].
Multiplexing Capabilities
The VirChip design accommodates 16-24 individual microchambers, enabling simultaneous detection of multiple pathogens from a single sample [37]. For LAMP-LFD approaches, multiplexing is achieved through target-specific probes with different haptens (digoxigenin, FITC) detected by corresponding antibodies on separate test lines [35].
Equipment Requirements and Simplification
True point-of-care application requires minimal equipment. The SPID platform uses a portable heating station rather than expensive thermocyclers [67]. For result interpretation, LFDs provide visual results without instrumentation, while smartphone-based readers can quantify fluorescence for more precise quantification [37] [35].
The integrated workflows presented herein demonstrate that complete LAMP-based diagnostic systems—from sample processing to result interpretation—can be implemented with minimal equipment while maintaining high sensitivity and specificity. These protocols provide researchers with validated methodologies for developing rapid viral diagnostics suitable for both clinical laboratories and point-of-care settings, advancing the field of molecular diagnostics toward truly decentralized testing.
Within the broader context of developing rapid viral diagnostics, high-throughput screening technologies are essential for managing large-scale infectious disease outbreaks and conducting extensive surveillance. Loop-mediated isothermal amplification (LAMP) has emerged as a particularly suitable molecular technique for this purpose. While standard LAMP reactions are celebrated for their simplicity and suitability in resource-limited settings, their adaptation to automated, high-throughput systems addresses a critical need for processing large volumes of samples efficiently in centralized laboratories. This application note details the establishment of semi-automated, high-throughput LAMP systems, providing validated protocols and performance data to enable researchers to implement these advanced screening solutions for robust, large-scale diagnostic applications [68].
Established High-Throughput LAMP Platforms
Recent advancements have led to the development of several LAMP-based systems designed to maximize throughput while minimizing manual intervention. These systems often leverage existing laboratory infrastructure to enhance processing capability and reproducibility.
Semi-Automated, Isolation-Free RT-LAMP for SARS-CoV-2
A significant demonstration of high-throughput LAMP was developed in response to supply bottlenecks during the COVID-19 pandemic [68]. This system successfully adapted a SARS-CoV-2 RT-LAMP protocol to a liquid handling station, significantly reducing manual hands-on time and processing errors. Key innovations included an isolation-free sample preparation using proteinase K digestion, which bypassed the need for RNA extraction from nasopharyngeal swabs. This approach, combined with automated liquid handling, facilitated the processing of 188 clinical samples with 100% sensitivity and specificity compared to standard RT-PCR [68]. The system utilized fluorescence detection on conventional RT-PCR cyclers, proving that high-throughput LAMP can be effectively deployed using instruments available in most clinical diagnostic laboratories.
Microplate-Based Colorimetric LAMP Detection
For laboratories requiring high-throughput, colorimetric LAMP analysis, a method has been developed to adapt tube-based assays to 384-well microplates monitored by a multimode microplate reader [69]. This platform kinetically measures the pH change inherent to nucleic acid amplification by tracking the ratio of 420 nm and 560 nm absorbance (420:560). The extensive DNA synthesis during LAMP releases hydrogen ions, acidifying the reaction mixture. Only true positive samples, confirmed through rigorous primer design, produce a significant increase in the 420:560 ratio, enabling automated, objective assessment of hundreds of reactions simultaneously [69]. Furthermore, operating this assay at an elevated temperature of 69 °C has been shown to reduce the time required to elicit positive reactions, thereby increasing throughput [69].
Table 1: Key Characteristics of High-Throughput LAMP Detection Platforms
| Platform Feature | Semi-Automated Fluorescence System | Microplate Colorimetric System |
|---|---|---|
| Detection Method | Fluorescence (probe-based) | Colorimetric (pH indicator) |
| Throughput Capacity | 188 samples per run [68] | 384-well plate format [69] |
| Automation Level | Liquid handling station [68] | Automated microplate reader [69] |
| Sample Preparation | Proteinase K digestion (isolation-free) [68] | Conventional or simplified DNA extraction |
| Reaction Time | ≤20 minutes [36] | ≤60 minutes [69] |
| Optimal Temperature | 63-65°C [68] | 65-69°C [69] |
Performance and Validation Data
High-throughput LAMP systems have demonstrated excellent performance characteristics in comparative studies, showing they are suitable for clinical diagnostic applications.
Comparative Sensitivity and Specificity
When validated against standard RT-PCR methods, the semi-automated SARS-CoV-2 RT-LAMP assay demonstrated an almost perfect agreement (Cohen's kappa > 0.8) with no systematic differences (McNemar's test, P > 0.05) [68]. The isolation-free protocol showed 100% sensitivity and 96.2% specificity compared to in-house RT-PCR, with a limit of detection (LoD) of 95 SARS-CoV-2 genome copies per reaction [68]. Similarly, a recent multi-platform LAMP system for detecting human adenovirus types 3 and 7 (HAdV-3 and HAdV-7) demonstrated 100% specificity with no cross-reactivity against SARS-CoV-2 or other respiratory pathogens [36].
Reproducibility and Quantitative Potential
The isolation-free SARS-CoV-2 RT-LAMP protocol showed high reproducibility, with an intra-run coefficient of variation (CV) for threshold time (Tt) of 0.4% and an inter-run CV of 2.1% [68]. A significant positive correlation (Rho > 0.8, P < 0.001) was observed between LAMP Tt values and RT-PCR Ct values, indicating the potential for semi-quantitative analysis [68]. The fluorescent probe LAMP method demonstrated superior sensitivity with an LoD of 1 copy/reaction and a median Ct value 72.8% lower than that of qPCR [36].
Table 2: Analytical Performance of High-Throughput LAMP Systems
| Performance Parameter | SARS-CoV-2 RT-LAMP (Isolation-Free) | HAdV LAMP (Multi-Platform) |
|---|---|---|
| Limit of Detection (LoD) | 95 copies/reaction [68] | 1 copy/reaction (Fluorescent probe method) [36] |
| Sensitivity | 100% (CI 91.0-100%) [68] | 100% (κ = 1.00; 95% CI: 1.00-1.00) [36] |
| Specificity | 96.2% (CI 80.4-99.9%) [68] | 100% (No cross-reactivity) [36] |
| Intra-Run Reproducibility | Tt CV = 0.4% [68] | Not specified |
| Inter-Run Reproducibility | Tt CV = 2.1% [68] | Not specified |
| Correlation with PCR | ϕ = 0.89, P < 0.001 [68] | Median Ct 72.8% lower than qPCR [36] |
Detailed Protocol: Semi-Automated High-Throughput LAMP
Equipment and Reagent Setup
Research Reagent Solutions:
- Bst 2.0 WarmStart DNA Polymerase (8,000 U/ml): Engineered for robust isothermal amplification with enhanced strand displacement activity [23]
- 2× RT-LAMP Premix 2.0 HS (Probe): Optimized ready-to-use mixture containing buffer, dNTPs, and magnesium for consistent high-sensitivity reactions [36]
- WarmStart RTx Reverse Transcriptase: Critical for reverse transcription in RNA virus detection, maintains activity at LAMP temperatures [35]
- LAMP Primers (F3, B3, FIP, BIP, LF, LB): Designed using PrimerExplorer V5 with HPLC purification to ensure specificity and minimize non-specific amplification [36] [70]
- Calcein or pH-Sensitive Dyes (phenol red): For colorimetric visualization; calcein changes from yellow to green, phenol red from magenta to yellow upon amplification [61] [69]
Automated Workflow Procedure
Sample Preparation (Isolation-Free Method):
- Transfer 50 μL of nasopharyngeal swab sample to a 96-well plate.
- Add 10 μL of proteinase K (20 mg/mL) to each sample.
- Incubate at 65°C for 10 minutes, then 98°C for 2 minutes to inactivate proteinase K.
- Centrifuge briefly to collect condensation [68].
Liquid Handler Setup:
- Program the liquid handling station to transfer 2.5 μL of processed sample to a 384-well PCR plate.
- Add 12.5 μL of 2× LAMP premix containing primers, dNTPs, and buffer.
- Include appropriate positive and negative controls in each run [68].
Amplification Protocol:
- Seal the plate and centrifuge briefly to ensure all content is at the bottom of wells.
- Incubate at 63°C for 30-40 minutes using a real-time PCR cycler with fluorescence acquisition every 30 seconds.
- For colorimetric detection, incubate in a thermal microplate reader at 65°C with kinetic measurement of 420:560 nm absorbance ratio [69].
Result Interpretation:
- Fluorescence method: Set a fluorescence threshold to determine Tt values; samples exceeding threshold within 20 minutes are positive.
- Colorimetric method: Samples showing a significant increase in 420:560 nm ratio are positive [69].
- Validation: Compare Tt values of samples to standard curve for semi-quantitation [68].
Technical Considerations for Implementation
Primer Design and Optimization
Successful high-throughput LAMP requires meticulous primer design. Primers should target 6-8 distinct regions of the target sequence, typically 15-25 bases in length with GC content of 40-60% [69]. Use dedicated software such as PrimerExplorer V5 or the NEB LAMP Primer Design Tool for initial design [70] [6]. Avoid runs of 3 or more identical bases or dinucleotide repeats. Outer primers (F3/B3) should have Tm values of 55-63°C, while internal and loop primers should have higher Tm values (60-68°C), with maximum difference of 5°C between all primers [69]. Always validate primer specificity using NCBI Primer-BLAST before implementation [36].
Troubleshooting Common Issues
- Non-Specific Amplification: Redesign primers using in silico tools to predict specificity and reduce primer-dimer formation [35]. Optimize Mg²⁺ concentration (typically 6-12 mM) and primer concentrations [23].
- Reduced Sensitivity in Automated System: Ensure liquid handler calibration is precise for small volumes. Include preheating steps to 65°C for all reagents to prevent non-specific amplification [69].
- Inconsistent Colorimetric Results: For pH-based detection, ensure consistent buffer capacity across all reaction components. Use fresh dye preparations and validate against fluorescent methods [69].
High-throughput LAMP systems represent a significant advancement in large-scale screening capabilities for viral diagnostics. The integration of semi-automated platforms with isolation-free sample preparation enables rapid processing of hundreds of samples with performance comparable to conventional RT-PCR. The availability of multiple detection formats—from fluorescence to colorimetric—allows laboratories to select the most appropriate platform based on their specific throughput needs and existing infrastructure. These systems effectively bridge the gap between rapid point-of-care testing and high-volume central laboratory diagnostics, making them invaluable tools for pandemic preparedness, routine surveillance, and large-scale research studies requiring efficient processing of numerous samples without compromising accuracy or reliability.
Optimizing LAMP Performance: Primer Design, Sensitivity Enhancement, and Contamination Control
Strategic Primer Design for Conserved Regions and Variant Coverage
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful molecular diagnostic tool for rapid viral detection, characterized by its isothermal nature, high efficiency, sensitivity, and specificity [1]. The technique employs 4 to 6 primers targeting 6 to 8 distinct regions of the target gene, enabling amplification at a constant temperature of 60-65°C and producing up to 10⁹ copies within an hour [1]. This application note details strategic primer design methodologies for targeting conserved genomic regions while maintaining coverage across viral variants, framed within the context of developing robust LAMP assays for rapid viral diagnostics.
The strategic design of primers is particularly crucial for viral diagnostics due to the high mutation rates of viruses and the emergence of variants that can evade conventional detection methods. By focusing on conserved regions while incorporating discriminatory elements for variant identification, researchers can develop assays that remain effective despite viral evolution. This approach has proven successful for detecting SARS-CoV-2 variants [71], hepatitis B virus [72], and various other viral pathogens.
Principles of LAMP Primer Design
Fundamental LAMP Mechanism
LAMP amplification utilizes a strand-displacing DNA polymerase and a set of specifically designed inner primers (FIP and BIP) and outer primers (F3 and B3) that recognize six distinct regions in the target DNA [1]. The reaction proceeds through the following mechanism:
- Initial Structure Formation: Inner primers FIP and BIP hybridize to the template and initiate synthesis, forming stem-loop structures through complementary F1c/F1 and B1c/B1 sequences.
- Strand Displacement: Outer primers F3 and B3 hybridize and displace the synthesized strands, releasing single-stranded DNA with loop structures at both ends.
- Cyclic Amplification: The dumbbell-shaped DNA structures serve as starting materials for continuous amplification, generating long DNA concatemers with alternating inverted repeats of the target sequence [1].
The addition of loop primers (LF and LB) has been shown to significantly accelerate the reaction by recognizing the loop regions formed during amplification, reducing reaction time from 60 to 30 minutes [71].
Conserved Region Selection Strategy
Identifying appropriate conserved regions requires comprehensive genomic analysis:
- Multi-Sequence Alignment: Collect and align target sequences from diverse variants and strains. For SARS-CoV-2 Delta variant detection, researchers analyzed sequences from the GISAID database and real-time mutation reports from outbreak.info [71].
- Conservation Scoring: Calculate conservation scores for each genomic region, prioritizing areas with minimal mutations across variants.
- Specificity Verification: Use BLASTN analysis against relevant databases to ensure the selected region is specific to the target pathogen [71].
- Functional Considerations: When possible, target genetically stable regions with essential biological functions, such as the nucleocapsid (N) gene in SARS-CoV-2, which demonstrates higher conservation than spike protein regions [71].
For hepatitis B virus detection, researchers designed pan-genotypic primer sets on conserved HBV gene regions that successfully detected eight major HBV genotypes/sub-genotypes (A1/2/3/B/C/D/E/F) with a detection limit ranging between 40 and 400 IU/mL [72].
Experimental Protocol for LAMP Assay Development
Primer Design Methodology
The following protocol outlines the complete workflow for designing and validating LAMP primers for conserved regions with variant coverage:
Step 1: Target Identification and Sequence Collection
- Identify target pathogens and specific variants of interest
- Download 15-25 representative sequences per target from databases (NCBI, GISAID)
- Select sequences based on length (preferably >600 bp) and diversity [6]
Step 2: Multiple Sequence Alignment
- Perform multiple sequence alignment using software such as Geneious Prime
- Generate consensus sequences (typically 300-400 bp) for each target organism [6]
- Identify regions of high conservation interspersed with variant-specific signatures
Step 3: Primer Design Using Specialized Tools
- Utilize dedicated LAMP primer design tools (NEB LAMP primer design tool)
- Design outer primers (F3, B3), inner primers (FIP, BIP), and loop primers (LF, LB)
- For variant discrimination, position the SNP at the 5' end of the loop primer [71]
Step 4: Specificity Validation
- Check primer specificity using blastn-short against NCBI database
- Verify minimal off-target binding through in silico analysis [6]
- For variant identification, ensure primers can discriminate single nucleotide polymorphisms
Step 5: Experimental Validation
- Synthesize primers and optimize reaction conditions
- Validate against reference samples with known variant status
- Determine sensitivity and specificity compared to reference methods
LAMP Reaction Setup
Reaction Composition:
- 10× Bst Buffer (200 mM Tris-HCl, 100 mM (NH₄)₂SO₄, 100 mM KCl, 20 mM MgSO₄, 1% Triton-100, pH 8.8)
- 1.6 mM dNTPs
- 8 mM MgSO₄
- 1.6 µM each of FIP and BIP primers
- 0.2 µM each of F3 and B3 primers
- 0.8 µM LF primer (and LB if applicable)
- 0.32 U/µL Bst DNA polymerase
- Template DNA/RNA
- Nuclease-free water to 25 µL [71]
Amplification Conditions:
- Incubate at 60-65°C for 30-60 minutes
- Enzyme activation at 95°C for 30 seconds (if using WarmStart enzymes)
- Reverse transcription at 60-65°C for 10-30 minutes for RNA targets [73]
Detection Methods and Results Interpretation
Detection Modalities
LAMP amplification products can be detected through multiple methods:
Turbidimetry: Measures white precipitate of magnesium pyrophosphate formed as a byproduct of amplification. Turbidity change correlates with amplified DNA quantity and can be monitored in real-time with turbidimeters [1].
Fluorometry: Uses DNA-intercalating dyes (SYTO-9, SYTO-13, SYTO-82, SYBR Green I, SYBR Gold, EvaGreen) that emit fluorescence when bound to double-stranded DNA. For SARS-CoV-2 detection, SYTO 9 provided excellent results in real-time fluorescence LAMP assays [71] [73].
Colorimetry: Enables visual detection without specialized equipment. Methods include:
- pH-sensitive indicators (xylenol orange) that change color from purple to yellow as pH decreases during amplification
- Metal ion indicators (calcein, eriochrome black T, hydroxy naphthol blue) that change color as Mg²⁺ levels decrease [1]
Molecular Beacons and Lateral Flow: Advanced detection methods providing enhanced specificity. Molecular beacons form hairpin structures with fluorophore and quencher that separate upon hybridization. Lateral flow assays enable rapid visual detection using tagged amplicons [10].
Quantitative Performance of LAMP Assays
Table 1: Analytical Performance of LAMP Assays for Various Pathogens
| Target Pathogen | Target Gene | Detection Limit | Time | Specificity | Reference |
|---|---|---|---|---|---|
| SARS-CoV-2 Delta variant | N gene (R203M) | Not specified | 30-50 min | 100% for Delta variant | [71] |
| Hepatitis B virus | Conserved HBV regions | 40-400 IU/mL | 60 min | 98.7% sensitivity, 91.5% specificity for ≥200,000 IU/mL | [72] |
| Aphanomyces euteiches (pea root rot) | ITS1 | 0.02 ng gDNA, 10 spores/sample | 60 min | High specificity against related species | [6] |
| Spirometra mansoni | cytb | 7.47 pg/μL (fecal DNA), 355.5 fg/μL (egg DNA) | Not specified | No cross-reaction with other common parasites | [74] |
| Diarrheagenic E. coli (STEC, EPEC, EHEC) | eae and stx2 | 10²–10³ gene copies/reaction | Not specified | Moderate specificity for eae and stx2 | [10] |
Variant Discrimination Strategies
For specific variant detection, the R203M mutation in the N gene of SARS-CoV-2 was successfully used as a Delta variant-specific marker. The genotyping RT-LAMP method was designed by analyzing the significant discrepancy in amplification efficiency between primers targeting the R203M-harboring region and primers targeting a conserved sequence of the N gene [71]. The Cq ratio between these two amplifications provided a reliable threshold (1.80) for distinguishing Delta from non-Delta variants with 100% accuracy [71].
This strategy can be adapted to other variants by identifying variant-specific SNPs in otherwise conserved regions and designing appropriate primer sets with the discriminatory base positioned to maximize amplification efficiency differences.
Research Reagent Solutions
Table 2: Essential Research Reagents for LAMP Assay Development
| Reagent/Kit | Function | Application Example | Key Features | |
|---|---|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase | General LAMP applications | 5'→3' polymerase activity, strand displacement capability, lacks 3'→5' exonuclease activity | [1] |
| Bst 2.0 & Bst 2.0 WarmStart | Enhanced DNA polymerase | Improved LAMP efficiency | Superior polymerization speed, thermal stability, salt tolerance, dUTP tolerance; WarmStart enables room temperature setup | [1] |
| Bst 3.0 | DNA polymerase with reverse transcriptase activity | RNA virus detection | Reverse transcriptase activity, robust amplification despite inhibitors, works with impure samples | [1] |
| SuperScript IV RT-LAMP Master Mix | Reverse transcription and amplification | SARS-CoV-2, measles, influenza detection | Optimized enzymatic reaction, operates at 65°C, 10-30 minute reaction time | [73] |
| Colorimetric ReadiLAMP SARS-CoV-2 Kit | Complete detection assay | SARS-CoV-2 surveillance | Colorimetric detection, direct LAMP option available, no sophisticated equipment needed | [73] |
| DNeasy PowerSoil Pro Kit | DNA extraction from complex samples | Soil and root samples for plant pathogens | Efficient DNA extraction from difficult matrices, inhibitor removal | [6] |
| QIAamp Viral RNA Mini Kit | Viral RNA extraction | Clinical samples for viral detection | High-quality RNA extraction, suitable for clinical specimens | [71] |
Workflow Visualization
LAMP Primer Design and Validation Workflow
LAMP Amplification Mechanism with Primer Binding
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful molecular diagnostic tool due to its isothermal nature, high speed, and robustness. For applications in rapid viral diagnostics, achieving a detection limit of 10-100 copies of the target nucleic acid is often critical for early infection detection and effective intervention. This sensitivity is influenced by a tightly interconnected set of factors including primer design, enzyme selection, reagent optimization, and detection methodology. This application note provides a detailed protocol and strategic framework for optimizing LAMP assays to consistently achieve this sensitivity target, framed within the context of advanced viral diagnostics research.
The Scientist's Toolkit: Essential Reagents for High-Sensitivity LAMP
The following reagents are fundamental for developing and executing a highly sensitive LAMP assay.
Table 1: Key Research Reagent Solutions for LAMP Optimization
| Reagent Category | Specific Examples | Function in LAMP Assay |
|---|---|---|
| DNA Polymerase | Bst 2.0 WarmStart, Bst 3.0 Polymerase [29] | Catalyzes DNA synthesis with strand-displacement activity essential for isothermal amplification. Bst 3.0 also possesses reverse transcriptase activity for direct RNA detection. |
| Primers | Inner (FIP, BIP), Outer (F3, B3), Loop (LF, LB) Primers [2] [26] | Specifically recognize 6-8 regions of the target sequence to ensure high specificity and efficiency. Loop primers accelerate the reaction, improving speed and sensitivity [29]. |
| dNTPs | dATP, dTTP, dCTP, dGTP [23] | The building blocks for DNA synthesis. Their concentration must be optimized for efficient amplification. |
| Magnesium Ions | MgSO₄ or MgCl₂ [23] | A critical cofactor for Bst DNA polymerase activity. Its concentration directly impacts enzyme efficiency and must be carefully optimized. |
| Detection Dyes | Hydroxynaphthol Blue (HNB), Calcein, SYBR Green I, EvaGreen [23] [29] | Enable result visualization. Metalochromic dyes like HNB signal amplification via magnesium depletion, while intercalating dyes fluoresce upon binding double-stranded DNA. |
Core Optimization Parameters and Experimental Protocols
Achieving a detection limit of 10-100 copies requires systematic optimization of several reaction components. The following data, synthesized from recent studies, provides a benchmark for optimization targets.
Table 2: Quantitative Optimization Targets for LAMP Assay Components
| Parameter | Typical Optimization Range | Recommended Target | Impact on Sensitivity |
|---|---|---|---|
| Reaction Temperature | 60 - 67 °C [23] | 63 - 65 °C [23] | Optimal Bst polymerase activity and primer binding specificity. |
| Reaction Time | 15 - 60 minutes [23] | 35 - 45 minutes [23] | Balances complete amplification of low-copy targets with preventing non-specific amplification. |
| Mg²⁺ Concentration | 6 - 12 mM [23] | 8 - 10 mM [23] | Critical cofactor for polymerase; insufficient Mg²⁺ reduces yield, excess can promote non-specificity. |
| dNTP Concentration | 1.0 - 1.6 mM [23] | 1.2 - 1.4 mM [23] | Adequate substrate supply for high-yield amplification. |
| Bst Polymerase | 6 - 12 Units/reaction [23] | 8 - 10 Units/reaction [23] | Sufficient enzyme to drive amplification from low-copy templates. |
| Inner Primers (FIP/BIP) | 0.8 - 3.2 µM [23] | 1.6 µM [23] | High local concentration to initiate strand invasion and cycling amplification. |
| Outer Primers (F3/B3) | 0.1 - 0.4 µM [23] | 0.2 µM [23] | Lower concentration than inner primers to facilitate slow strand displacement for loop formation. |
| Loop Primers (LF/LB) | 0.2 - 0.8 µM [23] | 0.4 µM [23] | Accelerate the reaction by binding loop regions, significantly reducing time-to-result. |
Detailed Optimization Protocol
This protocol outlines a step-by-step process for optimizing a LAMP assay for a novel viral target.
Experiment 1: Primer Design and In Silico Validation
- Target Selection: Identify a highly conserved region (e.g., the N gene in SARS-CoV-2) within the viral genome [26]. For RNA viruses, this region will be the target for reverse transcription.
- Primer Design: Use specialized software such as PrimerExplorer V5 to design a set of six primers: F3, B3, FIP, BIP, LF, and LB [23] [26]. The primers should target 6-8 distinct regions of the chosen sequence [2] [29].
- Specificity Check: Perform a BLAST analysis of all primer sequences against a genomic database (e.g., NCBI) to ensure specificity for the target virus and to avoid cross-reactivity with the host or co-infecting pathogens [23].
- In Silico Validation: Align all available sequences for the target viral strain (e.g., from GenBank) using tools like ClustalW to verify that the primer-binding regions are conserved across different variants [23] [26].
Experiment 2: Reaction Component Titration
- Prepare Master Mixes: Set up a series of LAMP reactions with a fixed amount of target DNA (e.g., 50 ng of a synthetic control plasmid) while varying the concentration of a single component (e.g., Mg²⁺) across its range (6, 8, 10, 12 mM). Keep other parameters constant [23].
- Amplification: Run the reactions at a constant temperature of 65 °C for 45 minutes.
- Analysis: Analyze the results using real-time turbidity measurement, fluorescence, or end-point gel electrophoresis to determine which condition yields the fastest amplification and highest yield.
- Iterate: Repeat steps 1-3 for each key component (dNTPs, polymerase, and each primer pair) sequentially. Use the optimal condition from one round as the new baseline for the next.
Experiment 3: Analytical Sensitivity and Limit of Detection (LOD) Determination
- Standard Preparation: Prepare a standard of known concentration. For absolute quantification, use a plasmid containing the target sequence. Calculate the copy number/μL using the formula: DNA copy number = [DNA concentration (g/μL) / (plasmid length in bp × 660)] × 6.02 × 10²³ [26].
- Serial Dilution: Perform a 10-fold serial dilution of the standard to create a concentration series spanning from 10⁸ down to 1 copy per reaction [26].
- LAMP Amplification: Run the optimized LAMP assay (from Experiment 2) with each dilution in replicate (n≥5).
- LOD Calculation: The LOD is defined as the lowest copy number at which ≥95% of the replicates test positive [26]. A properly optimized assay should reliably detect 10-100 copies.
The logical workflow for this multi-stage optimization process, from initial design to final validation, is summarized below.
Visualization and Detection Methods for Optimized Assays
The choice of detection method is crucial for interpreting the results of a high-sensitivity LAMP assay, especially in point-of-care contexts.
Table 3: Comparison of LAMP Product Detection Methods
| Method | Principle | Equipment Needed | Suitability for Field Use | Key Consideration |
|---|---|---|---|---|
| Colorimetric (Metalochromic Dyes) | Dye (e.g., HNB) changes color as Mg²⁺ is depleted during amplification [29]. | None (naked eye) | Excellent | Pre-amplification color (purple for HNB) changes post-amplification (sky blue). High contrast is critical for accurate visual interpretation [75]. |
| Colorimetric (pH Indicators) | Dye (e.g., Phenol Red) changes color with pH drop from amplification by-products [76]. | None (naked eye) | Excellent | Requires a weakly buffered reaction mixture to allow a detectable pH shift. |
| Turbidimetry | Measures white precipitate of magnesium pyrophosphate formed during amplification [29]. | Real-time turbidimeter | Moderate | Enables real-time, quantitative analysis. Not visualizable by naked eye alone. |
| Fluorometry | Fluorescent dye (e.g., SYBR Green I) intercalates into dsDNA and fluoresces [29]. | UV light or real-time fluorometer | Moderate to Low | Very high sensitivity. SYBR Green I is typically added post-amplification to prevent inhibition; closed-tube formats use specialized dyes. |
| Gel Electrophoresis | Separates DNA fragments by size, revealing a characteristic ladder pattern [23]. | Gel box, imager | No | Confirms specific amplification but is time-consuming and prone to amplicon contamination. |
The relationships and outcomes of different detection methods, particularly the straightforward colorimetric readout, can be visualized as follows.
The consistent achievement of a 10-100 copy detection limit in LAMP assays is attainable through a rigorous, systematic optimization process. This involves the strategic design of primers against conserved viral regions, meticulous titration of reaction components—particularly Mg²⁺, dNTPs, and primers—and the use of advanced polymerases like Bst 2.0 or 3.0. The optimized protocols outlined herein, validated by recent research, provide a reliable roadmap for researchers developing robust LAMP assays for sensitive viral detection. By integrating these optimized assays with field-suitable colorimetric detection, this framework supports the advancement of rapid, accurate, and accessible molecular diagnostics for viral diseases.
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful nucleic acid amplification technique, particularly valuable for rapid viral diagnostics in point-of-care and resource-limited settings [22]. Its advantages include operation at a constant temperature (60-65°C), rapid reaction times (often under 30-40 minutes), and minimal equipment requirements compared to conventional PCR [77] [22]. However, the technique's susceptibility to false-positive results presents a significant challenge for diagnostic reliability. These false positives primarily stem from primer-dimer formations, non-specific amplification, and aerosol contamination from amplicon carryover [78]. This application note provides detailed protocols and optimization strategies to mitigate these issues, enabling robust LAMP assay development within viral diagnostics research.
Core Principles and Causes of False-Positives
Primer-Dimer Formation
LAMP utilizes 4-6 primers recognizing 6-8 distinct regions of the target sequence, increasing the probability of primer-self annealing [22] [78]. Homodimers (between identical primers) and heterodimers (between different primers with complementary sequences) can form, serving as unintended templates for amplification and leading to false-positive signals, even in no-template controls (NTCs) [78].
Non-Specific Amplification
Non-specific amplification can occur when primers bind to non-target sequences with partial complementarity. The strand-displacing activity of Bst DNA polymerase can extend these misprimed complexes, generating amplification products in the absence of the true target [78].
Amplicon Contamination
The "cauliflower-like" structures and long concatemers generated by LAMP create substantial amplicon mass [77]. Opening reaction tubes for analysis risks aerosol contamination of laboratory surfaces and equipment, which can then contaminate subsequent reactions and cause false positives [78].
Optimization Strategies and Protocols
In Silico Primer Design and Validation
Principle: Meticulous primer design is the primary defense against false positives. LAMP primers (F3, B3, FIP, BIP, LF, LB) must be highly specific to the target viral sequence [22].
Protocol:
- Target Selection: Identify a highly conserved region within the viral genome. For SARS-CoV-2, the N gene (nucleotides 12-213) has been successfully targeted [26].
- Primer Design: Use specialized software such as PrimerExplorer V5 . Ensure primers meet the following criteria [26] [22]:
- Tm: FIP/BIP primers should be between 65°C and 70°C; F3/B3 primers should be 5-10°C lower.
- Length: F3/B3: 21-24 bp; FIP/BIP: 45-49 bp.
- GC Content: Maintain between 40% and 65%.
- Specificity: BLAST all primer sequences against relevant databases to ensure specificity.
- Validation: Assess primer sets for potential self-complementarity and dimer formation using tools like Primer-BLAST [78].
Wet-Lab Primer Testing and Optimization
Principle: Empirically determine the optimal primer concentration and ratio to minimize dimerization while maximizing sensitivity.
Protocol:
- Prepare Reaction Mix: Use a standard 25 µL reaction volume.
- Test Concentrations: Systematically vary primer concentrations. A suggested starting range is [23]:
- Outer Primers (F3/B3): 0.1 - 0.4 µM
- Inner Primers (FIP/BIP): 0.8 - 3.2 µM
- Loop Primers (LF/LB): 0.2 - 0.8 µM
- Analysis: Run reactions with NTCs. The optimal condition is the one that yields rapid amplification for positive templates and no signal in NTCs after 60 minutes. A study optimizing a sunflower mildew LAMP assay found 0.2 µM F3/B3, 1.6 µM FIP/BIP, and 0.4 µM LF/LB to be effective [23].
Reaction Condition Optimization
Principle: Fine-tuning physical and chemical reaction parameters enhances specificity.
Protocol:
- Temperature Gradient: Test amplification efficiency across a range (e.g., 63°C, 65°C, 67°C). Optimal temperature is often between 65°C [23].
- Additives: Include specificity-enhancing additives in the master mix.
- Hot-Start Techniques: Use advanced methods to prevent activity at lower temperatures.
- Gold Nanorods (AuNRs): Positively charged AuNRs electrostatically adsorb primers at room temperature, releasing them at ~50°C, which prevents mispriming. An assay for African horse sickness virus used this to achieve a detection limit of 100 copies/µL with high selectivity [79].
- Engineered Bst Polymerases: Use polymerases with inherent hot-start capability.
Contamination Control
Principle: Prevent amplicon carryover from contaminating new reactions.
Protocol:
- UDG Treatment: Incorporate dUTP in place of dTTP in the LAMP master mix. Add thermolabile Uracil-DNA Glycosylase (UDG). Any contaminating dU-containing amplicons from previous runs will be enzymatically degraded before amplification begins, while the target template (containing dT) remains unaffected. Incubate at 25°C for 5-10 minutes before the main LAMP reaction [77] [80].
- Spatial Separation: Physically separate pre- and post-amplification areas and use dedicated equipment and consumables [78].
Specific Detection Methods
Principle: Move beyond intercalating dyes to detection methods that confirm amplicon identity.
Protocol:
- Quenched Primers: Label LAMP primers (e.g., FIP) with a fluorophore that is quenched by guanine bases in close proximity within the amplicon. Specific amplification brings the fluorophore and guanine bases together, resulting in a measurable decrease in fluorescence, which is proportional to the starting template [81].
- Lateral Flow Assay (LFA): Design amplicons with an incorporated tag (e.g., FITC). After amplification, apply the product to an LFA strip. Tagged amplicons are captured by immobilized antibodies, providing a visual confirmation of specific target amplification [78].
Table 1: Summary of Key Optimization Parameters and Their Effects
| Parameter | Optimization Goal | Typical Range/Approach | Impact on False-Positives |
|---|---|---|---|
| Primer Concentration | Minimize dimerization | F3/B3: 0.1-0.4 µM; FIP/BIP: 0.8-1.6 µM [23] | High concentration increases primer interaction risk. |
| Reaction Temperature | Maximize specificity | 63-67°C [23] | Higher temperatures can enhance stringency. |
| Mg²⁺ Concentration | Balance enzyme efficiency | 6-12 mM [23] | Excess Mg²⁺ can promote non-specific amplification. |
| Additives (DMSO/Betaine) | Reduce secondary structures | DMSO: 1-5%; Betaine: 0.5-1.5 M [78] | Improves primer binding specificity. |
| Hot-Start Method | Prevent pre-incubation activity | AuNRs [79] or engineered Bst | Eliminates mispriming during setup. |
| Contamination Control | Degrade prior amplicons | UDG/dUTP system [77] [80] | Directly targets carryover contamination. |
Research Reagent Solutions
Table 2: Essential Reagents for Robust LAMP Assay Development
| Reagent / Material | Function / Rationale | Example Product / Note |
|---|---|---|
| Bst DNA Polymerase, LF | Strand-displacing enzyme core to LAMP. | Lyo-ready Bst (Thermo Fisher); Bst 2.0/3.0 (NEB); In-house purified WT Bst [41] [80] |
| Hot-Start Bst | Engineered to be inactive at room temperature. | Critical for reducing primer-dimer artifacts during reaction setup. |
| Thermolabile UDG | Enzymatically degrades contaminating dU-containing amplicons. | UDG BMTU (used in open-source assays) [80] |
| Reverse Transcriptase | For RT-LAMP to detect RNA viruses. | HIV-1 RT (open-source); WarmStart RTx [80] |
| Primer Design Software | Designs 4-6 primers for 6-8 target regions. | PrimerExplorer V5 [26] [22] |
| Specificity Additives | Chemicals that improve primer binding specificity. | DMSO, Betaine [78] |
| Gold Nanorods (AuNRs) | Provides a hot-start effect via electrostatic adsorption of primers. | Synthesized in-house with CTAB method [79] |
| Quenched Fluorescent Primers | Fluorophore-labeled primers for specific detection via quenching. | FAM-labeled FIP or LF primers [81] |
Workflow for False-Positive Reduction
The following diagram summarizes the integrated experimental workflow for developing a robust LAMP assay, from initial design to validation.
Reducing false positives in LAMP assays requires a multi-faceted strategy that begins with rigorous in-silico primer design and extends through wet-lab optimization of reaction conditions, the implementation of physical and enzymatic contamination controls, and the use of specific detection chemistries. By systematically applying the protocols and considerations outlined in this document, researchers can develop highly specific and robust LAMP assays, thereby unlocking the full potential of this powerful technology for reliable rapid viral diagnostics.
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful molecular diagnostic tool for rapid viral detection, particularly in point-of-care and resource-limited settings [82]. Its isothermal amplification conditions, which typically occur at 60-65°C, eliminate the need for sophisticated thermal cycling equipment required by conventional PCR methods [83] [31]. However, the technique faces a significant challenge: susceptibility to inhibitors present in clinical and environmental samples that can compromise assay sensitivity and specificity [84]. Non-specific amplification presents another frequent obstacle, often triggered by internal primers and primer-primer interactions at LAMP's relatively low reaction temperatures [83].
Effective inhibition mitigation requires a dual strategy addressing both sample processing and reaction chemistry. This application note provides detailed protocols and data-driven recommendations for identifying inhibition sources and implementing effective countermeasures to enhance LAMP assay robustness for viral diagnostics.
Understanding Inhibitory Substances and Their Mechanisms
Various substances encountered during sample collection, processing, or within sample matrices can inhibit LAMP amplification. Understanding these inhibitors is crucial for developing effective mitigation strategies.
Table 1: Common LAMP Inhibitors and Their Effects
| Inhibitory Substance | Common Sources | Effect on LAMP | Concentration Causing Inhibition |
|---|---|---|---|
| Humic Acid | Environmental samples, soil | Inhibits polymerase activity [84] | 4 ng/μL (with 50 ng DNA template) [84] |
| Melanin | Biological samples, tissues | Inhibits polymerase activity [84] | 0.05% (with 50 ng DNA template) [84] |
| EDTA (pH 6.5) | Lysis buffers, transport media | Chelates Mg²⁺ ions essential for Bst polymerase [84] | 10 mM [84] |
| Myoglobin | Blood, tissue samples | Binds to DNA or inhibits polymerase [84] | 4 μg/μL (with 50 pg DNA template) [84] |
| Urea | Clinical samples, urine | Disrupts hydrogen bonding [84] | 10 μg/μL (with 50 pg DNA template) [84] |
The inhibitory effects are often concentration-dependent and more pronounced when lower amounts of the target nucleic acid template are present [84]. Sample processing methods must therefore be optimized to remove or neutralize these substances before amplification.
Additive Strategies for Inhibition Mitigation
Chemical additives and proteins can enhance LAMP reaction robustness by suppressing non-specific amplification or counteracting the effects of inhibitors.
Additives for Suppressing Non-specific Amplification
Non-specific amplification remains a significant challenge in LAMP assay development, often requiring primer redesign or probe-based detection strategies. However, recent research has identified chemical additives that effectively mitigate this issue.
Table 2: Additives for Improving LAMP Specificity and Robustness
| Additive | Mechanism of Action | Optimal Concentration | Effectiveness |
|---|---|---|---|
| Tetramethylammonium chloride (TMAC) | Suppresses non-specific amplification by stabilizing DNA and preventing mispriming [83] | 20-60 mM [83] | Effectively suppresses non-specific amplification without primer redesign [83] |
| Dimethyl Sulfoxide (DMSO) | Reduces secondary structure formation in DNA and primers [83] | 2.5-7.5% (v/v) [83] | Moderately effective in specific conditions [83] |
| Formamide | Denatures secondary structures that cause mispriming [83] | 2.5-7.5% (v/v) [83] | Moderately effective in specific conditions [83] |
| Bovine Serum Albumin (BSA) | Binds to inhibitors; stabilizes enzymes [83] [84] | 0.1-0.5 mg/mL [83] | Reduces inhibition from humic acid, melanin, and urea [84] |
| T4 Gene 32 Protein (gp32) | Binds single-stranded DNA, prevents secondary structure [84] | Not specified in results | Reduces inhibition from humic acid, melanin, and urea [84] |
| Tween 20 | Reduces surface adsorption of enzymes [83] | 0.5-2% (v/v) [83] | Mild improvement in reaction consistency [83] |
Among these additives, TMAC has shown particular promise as a novel approach for suppressing non-specific amplification without the need for complex primer redesign or probe development [83]. The effectiveness of additives can be quantified by the difference in threshold cycles (ΔCt) between positive and negative samples, with higher ΔCt values indicating better suppression of false-positive signals [83].
Experimental Protocols
Protocol: Evaluating Additives for Inhibition Mitigation
This protocol outlines a systematic approach for testing and optimizing additives to overcome inhibition in LAMP assays.
Materials:
- WarmStart Bst 2.0 DNA Polymerase or equivalent
- Isothermal amplification buffer (10×, containing 2 mM MgSO₄ and 0.1% Tween 20)
- dNTP solution (10 mM each)
- Target primers (F3, B3, FIP, BIP, LF, LB)
- Additives: TMAC, DMSO, formamide, BSA, Tween 20
- Inhibitors: humic acid, melanin, myoglobin, urea, FeCl₃, EDTA
- DNA template (positive control)
- DEPC-treated water
- Thermocycler or heating block (65°C)
- Real-time fluorescence detection capability or colorimetric detection system
Method:
- Prepare inhibitor stocks: Create stock solutions of potential inhibitors at concentrations above expected inhibitory levels.
- Humic acid: 4 ng/μL and higher
- Melanin: 0.05% and higher
- EDTA: 10 mM (pH 6.5)
- Myoglobin: 4 μg/μL
- Urea: 10 μg/μL
Set up LAMP master mix (per reaction):
- 2.0 μL 10× isothermal amplification buffer
- 1.4 μL dNTP mix (10 mM each)
- 0.3 μL each of F3 and B3 primers (10 μM)
- 2.4 μL each of FIP and BIP primers (10 μM)
- 0.6 μL each of LF and LB primers (10 μM)
- 0.3-1.0 μL Bst DNA polymerase (8 U/μL)
- Variable: Additive(s) at test concentrations
- Variable: Inhibitor at test concentrations
- Nuclease-free water to 18 μL
Add template DNA:
- Add 2 μL of DNA template (positive control) or nuclease-free water (negative control) to reach a final reaction volume of 20 μL.
Amplification conditions:
- Incubate reactions at 60-65°C for 30-60 minutes.
- For real-time monitoring, record fluorescence every minute.
- For endpoint detection, use colorimetric methods (e.g., pH-sensitive dyes) or gel electrophoresis.
Data analysis:
- Calculate ΔCt values: ΔCt = Ct(negative) - Ct(positive)
- Higher ΔCt values indicate better specificity (effective suppression of non-specific amplification in negative controls)
- Compare amplification efficiency and time-to-positive across additive conditions
Protocol: Sample Processing to Minimize Inhibition
Proper sample processing is crucial for removing inhibitors prior to LAMP amplification, particularly when working with complex sample matrices.
Materials:
- Nasopharyngeal or oropharyngeal swabs
- Viral Transport Medium (VTM)
- Heating block or water bath (95°C)
- Centrifuge
- Proteinase K (optional)
- Commercial nucleic acid extraction kits (optional)
Method:
- Sample collection:
- Collect clinical specimen using appropriate swabs.
- Transfer swab to 2-3 mL of Viral Transport Medium (VTM) and agitate.
Thermal lysis:
- Aliquot 5-10 μL of VTM sample into a microfuge tube.
- Heat at 95°C for 1-5 minutes to lyse viral particles and inactivate nucleases.
- Briefly centrifuge to collect condensation.
Direct use or simplified extraction:
- Use 2-5 μL of heat-treated sample directly in LAMP reactions.
- For additional purification, consider simplified extraction methods:
- Proteinase K treatment (10-20 μg/μL, 56°C for 10 minutes, then 95°C for 5 minutes)
- Dilution of sample in nuclease-free water
- Brief centrifugation through silica columns if inhibitors persist
LAMP amplification:
- Incorporate optimized additives (e.g., 40 mM TMAC or 0.3 mg/mL BSA) into the LAMP master mix.
- Add processed sample to the reaction.
- Perform amplification at 65°C for 30-45 minutes.
- Monitor results in real-time or at endpoint.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for LAMP Inhibition Mitigation
| Reagent | Function | Application Notes |
|---|---|---|
| Tetramethylammonium chloride (TMAC) | Suppresses non-specific amplification | Most effective at 40 mM; enhances specificity without primer redesign [83] |
| Bovine Serum Albumin (BSA) | Binds inhibitors; stabilizes enzymes | Use at 0.1-0.5 mg/mL to counteract humic acid, melanin, and urea [84] |
| T4 Gene 32 Protein (gp32) | Single-stranded DNA binding protein | Reduces secondary structure; counteracts inhibitors [84] |
| WarmStart Bst 2.0 Polymerase | Strand-displacing DNA polymerase | Engineered for increased robustness against inhibitors [82] |
| WarmStart RTx Reverse Transcriptase | Reverse transcriptase for RT-LAMP | Enables RNA detection; included in master mixes for simplified setup [83] |
| SYBR Green I / Colorimetric Dyes | Detection methods | Enable real-time monitoring or visual readout; colorimetric dyes allow equipment-free detection [83] [82] |
Workflow and Mechanism Diagrams
Sample Processing and Inhibition Mitigation Workflow
LAMP Inhibition Causes and Mitigation Strategies
Temperature and Time Optimization for Maximum Efficiency
Loop-mediated isothermal amplification (LAMP) has emerged as a powerful molecular technique for rapid pathogen detection, offering significant advantages in speed, simplicity, and portability over conventional PCR methods. A critical factor determining the success of any LAMP assay is the precise optimization of reaction conditions, particularly incubation temperature and time. These parameters directly influence the activity of the strand-displacing DNA polymerase and the hybridization efficiency of the specially designed primer sets, ultimately affecting the assay's sensitivity, specificity, and speed. This Application Note provides a systematic framework for optimizing temperature and time parameters to achieve maximum efficiency in LAMP assays, with a specific focus on applications in viral diagnostics. The protocols and data presented herein form part of a broader thesis investigating the development of robust LAMP assays for rapid viral detection in both clinical and field settings.
The Scientist's Toolkit: Essential Reagents and Equipment
The following table catalogs key reagents and equipment essential for conducting LAMP optimization studies.
Table 1: Key Research Reagent Solutions for LAMP Assay Development
| Component | Function & Importance | Exemplary Products/Details |
|---|---|---|
| Strand-Displacing DNA Polymerase | Catalyzes DNA amplification at constant temperature; essential for isothermal reaction. | Bst 2.0 Warm Start DNA Polymerase [23]; Bst-XT WarmStart [85] |
| LAMP Primer Sets | Recognize 6-8 distinct regions of the target DNA; determine reaction specificity and efficiency. | Designed via PrimerExplorer V5 [23] [26] [86]; typically include F3, B3, FIP, BIP, and optional loop primers [86]. |
| Detection Reagents | Enable visualization of amplification results. | Colorimetric dyes (e.g., Neutral Red, Calcein) [23] [85]; fluorescent intercalators (e.g., SYBR Safe) [23]; pH-sensitive dyes [85]. |
| Isothermal Instrument | Maintains a constant temperature for the amplification reaction. | Simple heat blocks/water baths [23]; real-time thermal cyclers (e.g., Bio-Rad CFX96) [23]; portable field devices [85]. |
| Reaction Buffer | Provides optimal chemical environment (pH, salts) for polymerase activity. | Typically includes MgSO4, dNTPs, (NH4)2SO4, Tris-HCl, and Tween 20 [23]. |
Optimized Reaction Parameters: A Data Synthesis
Data aggregated from recent, high-quality studies on LAMP assay development reveal a consensus on effective temperature and time ranges. The following table summarizes optimized parameters for different applications, providing a reference point for assay development.
Table 2: Synthesis of Optimized Temperature and Time Parameters from Recent LAMP Studies
| Application Target | Optimal Temperature | Optimal Time | Key Performance Outcome | Source |
|---|---|---|---|---|
| Sunflower Mildew (Plasmopara halstedii) | 63°C, 65°C, 67°C | 15 - 60 min | Successful detection with DNA concentrations as low as 0.5 pg/μL. | [23] |
| SARS-CoV-2 (N Gene) | 58°C | 40 min | Limit of detection reached 10 copies/reaction. | [26] |
| Human Adenovirus (Hexon Gene) | 63°C | ≤ 20 min | Achieved a limit of detection of 1 copy/reaction with a fluorescent probe. | [36] |
| Nosocomial Pathogens (P. aeruginosa, S. aureus) | 65°C | 30 - 60 min | Demonstrated 100% sensitivity and specificity for S. aureus across all pre-incubation times. | [87] |
| SARS-CoV-2 (ORF1a Gene) | 62°C | 45 min | Showed high specificity and was deemed suitable for point-of-care testing. | [28] |
Experimental Protocol for Temperature and Time Optimization
This section provides a detailed, step-by-step methodology for empirically determining the optimal temperature and time for a new LAMP assay.
Materials and Reagents
- Purified target DNA (or RNA with RT-LAMP components)
- 2X LAMP Master Mix (e.g., containing Bst polymerase, dNTPs, and reaction buffer)
- Designed LAMP primer set (FIP, BIP, F3, B3, LF, LB)
- Nuclease-free water
- MgSO4 (if not included in master mix at optimal concentration)
- Real-time isothermal thermal cycler or heated block with precise temperature control
- Tube strips or plates compatible with the detection instrument
Procedure
- Reaction Mixture Preparation: Prepare a master mix on ice containing per reaction: 12.5 μL of 2X LAMP Master Mix, 1.6 μM each of FIP and BIP primers, 0.2 μM each of F3 and B3 primers, 0.4 μM each of LF and LB primers [23] [88], and a predetermined optimal concentration of Mg2+ (e.g., 6-8 mM) [23]. Adjust the final volume to 23 μL/reaction with nuclease-free water.
- Template Addition: Aliquot 23 μL of the master mix into each reaction tube. Add 2 μL of template DNA (e.g., 50 ng/reaction for optimization [23]) to the test reactions. Include a no-template control (NTC) containing 2 μL of nuclease-free water to monitor for contamination.
- Temperature Gradient Experiment:
- Program the thermal cycler for a set time (e.g., 45 minutes) and run identical reactions across a temperature gradient. A recommended range is 58°C to 67°C [23] [26] [87].
- If using a real-time instrument, monitor fluorescence continuously. If using an end-point method, ensure the reaction is stopped simultaneously for all temperatures.
- Time-Course Experiment:
- Data Analysis:
- For real-time data: The optimal condition is identified by the shortest time to positive (Tp) and the highest amplification efficiency (steepest curve slope) [36].
- For end-point data (e.g., gel electrophoresis, colorimetry): The optimal condition is the shortest time that yields a strong, specific amplification signal with a negative NTC.
Figure 1: A sequential workflow for the systematic optimization of temperature and time in a LAMP assay.
Discussion and Concluding Remarks
The optimization of temperature and time is a foundational step in developing a robust LAMP assay. Data from recent studies consistently indicate that a temperature range of 58–67°C is effective for most targets, with many assays converging around 63–65°C [23] [87]. The optimal temperature is a balance that facilitates efficient primer binding and strand displacement while maintaining enzyme stability.
The reaction time is intrinsically linked to the target concentration, primer efficiency, and reaction temperature. While some assays can yield results in as little as 15–20 minutes [23] [36], a 45–60 minute incubation often ensures high sensitivity, especially for low-abundance targets. The use of loop and swarm primers can significantly reduce the time to result by increasing the number of initiation sites for DNA synthesis [86].
A key strength of LAMP is its compatibility with multiple detection methods. The optimized protocols can be adapted for real-time fluorescence, colorimetric (pH-sensitive or metal indicator) dyes, or lateral flow immunochromatography, making the technology versatile for laboratory, point-of-care, and field use [36] [85]. The optimized conditions detailed in this note provide a reliable foundation for establishing sensitive, rapid, and reliable LAMP assays suitable for the demanding needs of modern viral diagnostics research.
Probe Incorporation and Endonuclease Integration for Enhanced Specificity
Loop-mediated isothermal amplification (LAMP) is a powerful molecular diagnostic technique renowned for its rapidity, sensitivity, and operational simplicity, making it particularly suitable for point-of-care and resource-limited settings [61]. The method employs a DNA polymerase with high strand displacement activity and four to six primers that recognize six to eight distinct regions on the target DNA, which theoretically confers a high degree of specificity [61] [89]. Amplification occurs at a constant temperature (typically 60–65°C) in less than an hour, eliminating the need for sophisticated thermocycling equipment [61]. Despite these advantages, a significant drawback of conventional LAMP is its frequent reliance on indirect detection methods, such as turbidity measurement or intercalating dyes (e.g., SYBR Green I), which detect amplification byproducts rather than the target sequence itself [61] [89]. This reliance poses a substantial risk of false-positive results due to non-specific amplification, often arising from primer-dimer formations or mis-priming events [61] [89]. Such limitations can severely compromise diagnostic accuracy, especially in clinical settings.
To address these challenges, this Application Note focuses on advanced strategies that integrate sequence-specific probes and endonuclease enzymes into the LAMP workflow. These approaches transform LAMP from a non-specific amplification method into a highly specific nucleic acid detection system, enabling real-time monitoring and significantly reducing the potential for false-positive outcomes, thereby enhancing its reliability for critical viral diagnostic applications.
Probe-Based Detection Strategies
The incorporation of target-specific probes into LAMP assays provides a direct mechanism for detecting amplification products, thereby overcoming the inherent non-specificity of dye-based methods.
Self-Dequenching Fluorogenic Probes (FLOS-LAMP)
The FLOS-LAMP (Fluorescence of Loop Primer Upon Self Dequenching-LAMP) technology utilizes oligonucleotide probes labelled with a fluorophore that is quenched in the unbound state due to its proximity to a guanine base or other quenching moieties in the sequence [89]. Upon hybridization to the complementary target sequence within the LAMP amplicon, the probe undergoes a conformational change that disturbs the quenching interaction, resulting in a detectable fluorescent signal [89].
- Probe Design and Mechanism: The probe is typically designed to bind to the loop region of the LAMP amplicon. In its free, unbound form, the fluorophore's fluorescence is suppressed. When the probe hybridizes to its specific target loop during amplification, the fluorophore is separated from the quencher, leading to de-quenching and a significant increase in fluorescence that can be monitored in real-time [89].
- Performance and Advantages: This method offers direct, sequence-specific confirmation of the target amplicon. It has been successfully validated for detecting Varicella-zoster virus (VZV), demonstrating a limit of detection (LOD) of 500 copies and high clinical sensitivity (96.8%) and specificity (100%) [89]. The key advantage is the drastic reduction of false positives, as signal generation is contingent upon specific probe hybridization, not just the presence of any double-stranded DNA [89].
Fluorescent Hydrolysis Probes
This approach adapts the principles of TaqMan probe chemistry for LAMP, utilizing a dual-labelled probe (e.g., with HEX and BHQ1) that is cleaved by the endonuclease activity of the Bst DNA polymerase during amplification.
- Probe Design and Mechanism: A probe is designed to be complementary to an internal sequence of the LAMP amplicon. The 5' end is labelled with a reporter fluorophore, and the 3' end is labelled with a quencher. When intact, the proximity of the quencher suppresses the reporter's fluorescence. During the strand displacement process of LAMP, the Bst polymerase cleaves the bound probe, separating the fluorophore from the quencher and resulting in a permanent fluorescent signal increase [36].
- Performance and Advantages: This method is highly sensitive and specific. A study on Human Adenovirus (HAdV) detection reported that the fluorescent probe method demonstrated a superior LOD of 1 copy/reaction, which was more sensitive than associated qPCR assays (median Ct value of 7.3 for LAMP vs. 26.9 for qPCR) [36]. It also showed 100% specificity with no cross-reactivity against other respiratory pathogens [36]. The real-time fluorescence monitoring allows for quantitative analysis.
Table 1: Comparison of Probe-Based LAMP Detection Methods
| Feature | FLOS-LAMP (Self-Dequenching) | Fluorescent Hydrolysis Probes |
|---|---|---|
| Probe Structure | Single-labeled oligonucleotide | Dual-labeled (fluorophore-quencher) oligonucleotide |
| Signal Mechanism | De-quenching upon hybridization | Fluorophore release via endonuclease cleavage |
| Key Enzyme | Bst DNA Polymerase (strand displacement) | Bst DNA Polymerase (strand displacement + endonuclease) |
| Real-time Capability | Yes | Yes |
| Reported Sensitivity | 500 copies (VZV) [89] | 1 copy/reaction (HAdV) [36] |
| Reported Specificity | 100% (VZV) [89] | 100% (HAdV) [36] |
Experimental Protocol: Real-Time LAMP with Fluorescent Hydrolysis Probes
The following protocol is adapted from a multi-platform HAdV detection system [36] and is designed for a 25 µL reaction volume.
Research Reagent Solutions:
- Bst DNA Polymerase: A strand-displacing DNA polymerase (e.g., Bst 2.0 Warm Start) with optional endonuclease activity for probe cleavage.
- LAMP Primers: A set of six primers (F3, B3, FIP, BIP, LF, LB) targeting the pathogen of interest.
- Dual-Labelled Probe: A hydrolysis probe (e.g., 5'-HEX, 3'-BHQ1) complementary to the target sequence within the LAMP amplicon.
- Reaction Buffer: Isothermal amplification buffer, typically containing MgSO₄, (NH₄)₂SO₄, Tris-HCl, and Tween 20.
- dNTPs: Deoxynucleotide solution.
- Nucleic Acid Template: Extracted DNA or RNA (for RT-LAMP, adding a reverse transcriptase).
Procedure:
- Reaction Mix Preparation: On ice, combine the following components in a PCR tube:
- 12.5 µL of 2x LAMP Premix (commercial or prepared in-house)
- 1.0 µL of Primer Mix (containing 8 µM each of F3 and B3, 48 µM each of FIP and BIP, and 16 µM each of LF and LB) [90]
- 1.0 µL of Dual-Labelled Probe (5 µM stock concentration)
- 2.0 µL of DNA template (or 5 µL for RNA in RT-LAMP)
- Nuclease-free water to a final volume of 25 µL
- Instrument Setup: Place the tubes in a real-time fluorometer or thermal cycler with isothermal capability. Set the fluorescence channel to match the reporter dye (e.g., HEX).
- Amplification: Run the reaction at 63°C for 30-45 minutes, with fluorescence acquisition at 1-minute intervals [36] [90].
- Data Analysis: Determine positive results based on the time to positivity (Tp) or the fluorescence curve crossing a predetermined threshold. No-template and negative template controls are essential for validation.
Diagram 1: Hydrolysis probe mechanism workflow.
Quantitative Performance of Enhanced LAMP Assays
The integration of probe-based detection and endonuclease activity significantly improves the analytical performance of LAMP assays, making them comparable to, and in some cases surpassing, traditional qPCR.
Table 2: Analytical Performance of Advanced LAMP Platforms
| Pathogen Detected | LAMP Platform | Limit of Detection (LOD) | Clinical Sensitivity | Clinical Specificity |
|---|---|---|---|---|
| Human Adenovirus (HAdV) [36] | Fluorescent Probe LAMP | 1 copy/reaction | 100% | 100% |
| Human Adenovirus (HAdV) [36] | Calcein/IC LAMP | 2.5 copies/reaction | 100% | 100% |
| Varicella-Zoster Virus (VZV) [89] | FLOS-LAMP | 500 copies | 96.8% | 100% |
| Avian Influenza Virus (AIV) [91] | Real-Time RT-LAMP (SYBR Green) | Ct = 38 (vs. RT-PCR) | 100% | 100% |
| Avian Influenza Virus (AIV) [91] | Colorimetric LAMP | Ct = 32 (vs. RT-PCR) | 91.67% | 100% |
| Clostridium perfringens [90] | CP-LAMP (cpa gene) | Comparable to PCR | 100% | 88.33% - 100% |
The data in Table 2 underscore the high sensitivity and specificity achievable with advanced LAMP methods. The fluorescent probe LAMP for HAdV is particularly noteworthy for its single-copy sensitivity, which outperformed a commercial qPCR kit in a clinical validation study [36]. Similarly, the FLOS-LAMP assay for VZV demonstrated excellent clinical performance, validating the utility of probe-based specificity in a diagnostic context [89].
The Scientist's Toolkit: Essential Reagents for Probe-Based LAMP
Successful implementation of these advanced LAMP protocols requires a set of core reagents, each playing a critical role in the reaction's efficiency and specificity.
Table 3: Research Reagent Solutions for Enhanced LAMP Assays
| Reagent | Function | Example Specifications / Notes |
|---|---|---|
| Bst DNA Polymerase | Catalyzes isothermal DNA amplification; some versions possess endonuclease activity for probe cleavage. | Bst 2.0 or 3.0 Warm Start; 8,000 U/mL concentration; use 6-12 U per reaction [23] [90]. |
| LAMP Primers | Specifically bind 6-8 target regions to initiate strand-displacement amplification. | F3/B3 (0.1-0.4 µM), FIP/BIP (0.8-3.2 µM), LF/LB (0.2-0.8 µM) [23]. HPLC purification recommended. |
| Fluorescent Probes | Provide sequence-specific detection via hybridization or hydrolysis. | For FLOS-LAMP: Single-labeled probe (e.g., FAM). For Hydrolysis: Dual-labeled (e.g., 5'-HEX/3'-BHQ1) [36] [89]. |
| Reaction Buffer | Provides optimal pH, ionic strength, and co-factors for polymerase activity. | Typically contains MgSO₄ (6-12 mM), (NH₄)₂SO₄, Tris-HCl, KCl, and Tween 20 [23] [91]. |
| dNTPs | Building blocks for new DNA strand synthesis. | Use at 1.0-1.6 mM final concentration in the reaction [23]. |
| Reverse Transcriptase | For RT-LAMP, converts target RNA into cDNA for amplification. | Can be included in the reaction mix for one-step RT-LAMP [91]. |
Diagram 2: Key reagent functional relationships.
The integration of probe-based detection systems and endonuclease activity into LAMP assays marks a significant advancement in molecular diagnostics. The FLOS-LAMP and fluorescent hydrolysis probe methods detailed herein effectively address the critical challenge of non-specific amplification, transforming LAMP into a highly specific and reliable quantitative tool. The provided protocols and performance data demonstrate that these enhanced LAMP assays can achieve sensitivity and specificity on par with, and sometimes exceeding, gold-standard qPCR methods. This, combined with LAMP's inherent advantages of speed, isothermal conditions, and minimal equipment requirements, positions probe-based LAMP as a formidable platform for rapid and accurate viral diagnostics in both laboratory and point-of-care settings.
LAMP Assay Validation: Clinical Performance Metrics and Comparative Analysis with Gold-Standard Methods
Clinical Sensitivity and Specificity Assessment in Patient Cohorts
Loop-mediated isothermal amplification (LAMP) has emerged as a critical molecular technology for the rapid diagnosis of infectious diseases in clinical settings. As a nucleic acid amplification technique, LAMP operates at a constant temperature (typically 60-65°C) and utilizes a strand-displacing DNA polymerase with 4-6 specific primers that recognize distinct regions of the target DNA, enabling highly specific amplification without the need for thermal cycling equipment [92]. This technical profile makes LAMP particularly suitable for resource-limited environments and point-of-care testing scenarios where rapid turnaround times are essential for clinical decision-making. The assessment of clinical sensitivity and specificity in well-characterized patient cohorts represents a fundamental step in validating LAMP assays before implementation in diagnostic pathways, ensuring reliable performance across diverse clinical specimens and patient populations.
This Application Note provides a comprehensive framework for evaluating LAMP assay performance in clinical research settings, with structured methodologies for determining sensitivity and specificity parameters against reference standards. The protocols and data analysis approaches outlined here are designed to support researchers in generating robust evidence for the diagnostic accuracy of LAMP assays across various infectious disease targets, facilitating their translation from research tools to clinically implemented diagnostics.
Table 1: Diagnostic Performance of LAMP Assays Across Pathogens and Sample Types
| Pathogen | Sample Type | Clinical Sensitivity (%) | Clinical Specificity (%) | Reference Standard | Study/Context |
|---|---|---|---|---|---|
| Mycobacterium tuberculosis | Respiratory specimens | 84.1 (95% CI: 78.3-88.6) | 96.1 (95% CI: 94.2-97.4) | Microbiological or composite reference standard | Pulmonary tuberculosis detection [93] |
| Mycobacterium tuberculosis | Lymph node tissue | 94.3 (95% CI: 79.8-98.6) | 90.0 (95% CI: 79.5-95.4) | Microbiological or composite reference standard | Lymph node tuberculosis [93] |
| Streptococcus pneumoniae | Sterile body fluids (pleural fluid, plasma, CSF) | 100.0 | 99.3 | Real-time PCR | Invasive pneumococcal disease [94] |
| SARS-CoV-2 | Nasopharyngeal swabs (RNA-LAMP) | 92.91 | 98.33 | RT-PCR | COVID-19 diagnosis [95] |
| SARS-CoV-2 | Nasopharyngeal swabs (Direct LAMP) | 70.92 | 99.86 | RT-PCR | COVID-19 diagnosis [95] |
| SARS-CoV-2 | Pharyngeal swabs | 87.0 (97.0 for Ct<35) | 98.0 | RT-PCR | Multicenter field trial [96] |
| Foodborne bacteria | Various food samples | 96.6 (95% CI: 95.0-97.7) | 97.6 (95% CI: 92.6-99.3) | Culture method | Meta-analysis [92] |
| Leptospira spp. | Clinical samples | 94.0 (95% CI: 86.0-97.0) | 96.0 (95% CI: 94.0-98.0) | PCR and other standards | Meta-analysis [97] |
The performance metrics summarized in Table 1 demonstrate that well-optimized LAMP assays consistently achieve high sensitivity and specificity across diverse pathogen targets and sample matrices. The technology shows particular strength in detecting pulmonary tuberculosis with 84.1% sensitivity and 96.1% specificity [93], and exhibits excellent performance for invasive pneumococcal disease with 100% sensitivity and 99.3% specificity compared to PCR [94]. The data also reveal that sample processing methodology significantly impacts performance, as evidenced by the substantial difference in sensitivity between RNA-LAMP (92.91%) and direct LAMP (70.92%) for SARS-CoV-2 detection [95].
LAMP assays perform particularly well in patient cohorts with higher pathogen loads, as demonstrated by the superior sensitivity for SARS-CoV-2 in samples with Ct values <35 (97%) compared to those with higher Ct values (60%) [96]. This performance characteristic positions LAMP as an excellent tool for early infection detection when viral or bacterial loads are typically higher, or for diagnosing diseases where pathogen burden correlates with clinical severity.
Experimental Protocol for Sensitivity and Specificity Assessment
Patient Cohort Selection and Sample Collection
Objective: To establish a representative patient cohort for evaluating clinical sensitivity and specificity of LAMP assays.
Materials:
- Appropriate sample collection kits (swabs, sterile containers, transport media)
- Ethical approval documentation
- Clinical data collection forms
- Freezer facilities for sample storage (-70°C to -80°C recommended)
Procedure:
- Define Inclusion/Exclusion Criteria: Establish clear clinical criteria for patient enrollment, including symptomatic presentation, exposure history, and demographic considerations. For respiratory pathogen detection, enroll patients with clinically suspected infection based on symptoms (e.g., cough, fever, dyspnea) and radiological findings [64].
- Obtain Ethical Approval: Secure approval from institutional review boards before study initiation. For the tuberculosis LAMP study, WHO provided ethical oversight [93].
- Collect Clinical Specimens: Obtain appropriate samples based on syndrome:
- For pulmonary tuberculosis: Collect respiratory specimens (sputum, bronchoalveolar lavage) [93]
- For invasive pneumococcal disease: Collect sterile body fluids (CSF, blood, pleural fluid) [94]
- For hospital-acquired pneumonia: Collect bronchoalveolar lavage, endotracheal aspirates [64]
- Record sample quality metrics (e.g., Murray-Washington criteria for respiratory samples) [64]
- Process Samples: Aliquot samples for parallel testing by LAMP and reference methods. Process samples within 2 hours of collection or store according to validated conditions [64].
- Collect Clinical Data: Document patient demographics, symptom onset, treatment history, and other relevant clinical parameters.
Nucleic Acid Extraction and LAMP Assay Procedure
Objective: To isolate target nucleic acids and perform LAMP amplification for pathogen detection.
Materials:
- Nucleic acid extraction kit (e.g., magnetic bead-based system)
- LAMP primer sets (4-6 primers per target)
- LAMP reaction mix (including buffer, betaine, MgSO4, dNTPs)
- Bst DNA polymerase with strand displacement activity
- Isothermal amplification instrument (e.g., Genie II, conventional heat block, or real-time PCR system)
- Optional: Visual detection additives (calcein, hydroxy naphthol blue, or pH-sensitive dyes)
Procedure:
- Nucleic Acid Extraction:
LAMP Reaction Setup:
- Prepare LAMP master mix containing:
- 2.5-5.0μL 10× isothermal amplification buffer
- 1.5-2.5μL MgSO4 (6-8 mM final concentration)
- 3.5-4.5μL betaine (0.8-1.2 M final concentration)
- 1.0-1.5μL dNTP mix (1.0-1.4 mM each)
- 0.5-1.5μL primer mix (0.08-0.2 μM F3/B3, 0.8-2.0 μM FIP/BIP, 0.4-1.0 μM LF/LB)
- 0.5-1.5μL Bst DNA polymerase (8-16 units)
- Nuclease-free water to final volume
- Aliquot 20-25μL master mix into reaction tubes
- Add 5-10μL template DNA
- Include appropriate controls (positive, negative, no-template)
- Prepare LAMP master mix containing:
Amplification and Detection:
- Incubate reactions at 60-67°C for 30-90 minutes
- For real-time detection, monitor fluorescence every 30-60 seconds
- For visual detection, add colorimetric dyes before or after amplification
- For lateral flow detection, use biotin- and FAM-labeled primers [36]
Result Interpretation:
- For real-time systems: Use threshold time or Ct-equivalent values
- For visual detection: Assess color change (e.g., from pink to yellow for phenol red)
- Establish cutoff values based on validation studies
Reference Testing and Discrepancy Analysis
Objective: To compare LAMP results against reference standards and resolve discordant findings.
Materials:
- Reference standard reagents (culture media, PCR kits)
- Discrepancy testing materials (alternative molecular methods, sequencing)
Procedure:
- Perform Reference Testing:
Discrepancy Analysis:
- Retest discordant samples with alternative molecular method
- Use sequence analysis for definitive confirmation
- For LAMP-positive/culture-negative samples, consider higher sensitivity of molecular methods
- For LAMP-negative/culture-positive samples, investigate inhibition or target mismatch
Data Analysis:
- Calculate sensitivity, specificity, PPV, NPV with 95% confidence intervals
- Perform stratified analysis by sample type, pathogen load, symptom duration
- Assess inter-reader variability for visual detection methods
Research Reagent Solutions
Table 2: Essential Reagents and Materials for LAMP Assay Development
| Reagent/Material | Function | Examples/Specifications |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase for isothermal amplification | Bst 2.0/WarmStart Bst (8-16 units/reaction) [94] |
| LAMP Primers | Specific recognition of 6-8 target regions | F3, B3 (0.08-0.2 μM); FIP, BIP (0.8-2.0 μM); LF, LB (0.4-1.0 μM) [94] |
| Detection Reagents | Visual or fluorescent signal generation | Calcein, hydroxy naphthol blue, SYBR Green, intercalating dyes [36] [98] |
| Nucleic Acid Extraction Kits | Isolation of DNA/RNA from clinical samples | Magnetic bead-based systems (e.g., NucliSENS EasyMag) [94] |
| Isothermal Instruments | Constant temperature amplification | Genie II, conventional heat blocks, real-time PCR systems [64] |
| Modified Nucleotides | Amplicon labeling for detection systems | Biotin-dUTP, Cy5-dUTP, aminoallyl-dUTP for lateral flow or microarray detection [98] |
| Sample Processing Reagents | Sample preparation and liquefaction | Sputum liquefying solutions (e.g., DTT-containing solutions) [64] |
Workflow Diagram for Clinical Validation
Figure 1: Clinical validation workflow for LAMP assays, illustrating the sequential steps from study design through result interpretation, with parallel testing against reference standards.
Critical Implementation Considerations
Sample Processing Optimization
Sample preparation methodology significantly impacts LAMP assay performance. For respiratory samples, pre-treatment with liquefying agents such as dithiothreitol (DTT) is essential for viscous specimens like endotracheal aspirates and bronchoaspirates [64]. The dilution factor must be optimized to balance inhibition removal with target concentration. For direct LAMP methods without nucleic acid extraction, heat inactivation at 95°C for 5 minutes provides adequate sample preparation while maintaining simplicity [95]. However, the substantial difference in sensitivity between RNA-LAMP (92.91%) and direct LAMP (70.92%) for SARS-CoV-2 detection indicates that extraction-based methods generally provide superior clinical sensitivity [95].
Inhibition Control Strategies
Inhibition represents a significant challenge in molecular diagnostics from complex clinical matrices. The high tolerance of Bst polymerase to inhibitory substances present in biological samples provides LAMP with an advantage over PCR in some applications [94]. Nevertheless, incorporation of internal amplification controls is essential to distinguish true negative results from inhibition. For quantitative interpretation, standard curves using external controls spiked into negative clinical matrix can monitor inhibition effects [94]. For qualitative tests, non-competitive internal controls can validate each individual reaction.
Multiplexing Approaches
Multiplex LAMP detection presents technical challenges due to the high number of primers required (4-6 per target) and associated risk of primer-dimer formations and non-specific amplification [64]. Strategies for successful multiplexing include careful primer design using specialized software, empirical optimization of primer ratios, and incorporation of probe-based detection systems [36]. When developing multiplex panels, balance the number of targets against potential sensitivity reduction, as assays with fewer targets typically achieve higher sensitivity and specificity [64].
Quality Assurance Framework
Robust quality management is essential for clinical implementation of LAMP assays. Establish acceptance criteria for control samples, including positive control time-to-positive ranges and negative control maximum fluorescence thresholds [94]. For visual detection methods, implement blinded reading by multiple operators to assess inter-reader variability [95]. Document lot-to-lot reagent variation through validation of new reagent batches against retained positive clinical samples. Regular participation in external quality assessment programs provides critical performance verification for clinical laboratory implementation.
Within the broader scope of thesis research on Loop-Mediated Isothermal Amplification (LAMP) for rapid viral diagnostics, a critical evaluation of its reliability against the gold standard, reverse transcription quantitative polymerase chain reaction (RT-qPCR), is paramount. This Application Note synthesizes empirical evidence from multiple studies to provide a detailed comparison of these methodologies, focusing on concordance rates and the nature of analytical discrepancies. The objective is to furnish researchers, scientists, and drug development professionals with a clear framework for validating LAMP assays, ensuring their deployment in clinical, agricultural, and public health settings is both robust and reliable. The data and protocols herein are designed to support the experimental phase of thesis work, providing a benchmark for performance evaluation.
Quantitative Concordance Analysis
Direct comparisons between LAMP and RT-qPCR reveal a high degree of concordance, though performance is influenced by the specific pathogen, primer sets, and sample matrix. The table below summarizes key findings from several studies to facilitate a direct quantitative comparison.
Table 1: Direct Comparison of LAMP and RT-qPCR Assay Performance
| Pathogen / System | Sample Type | LAMP Assay Details | Concordance with RT-qPCR | Key Performance Metrics | Citation |
|---|---|---|---|---|---|
| SARS-CoV-2 | Clinical RNA samples | 10 different RT-LAMP primer sets | 44.8% - 82.8% (Positive Detection Rate) | Set-4 (Nsp3 target) showed highest detection rate (82.8%); Set-14 (N target) showed fastest amplification (Tt < 8.5 min). | [99] |
| SARS-CoV-2 | Contrived clinical samples | 3 commercial RT-LAMP kits | Kappa (κ): 0.75 - 0.93 | Clinical sensitivity varied by kit; specificity was consistently high. | [100] |
| Pea Root Rot Pathogens | Soil and infected root tissue | ITS1-targeted LAMP | Comparable or superior to qPCR | Limit of Detection (LOD): 0.02 ng gDNA or 10 spores per sample. Results within 60 min. | [6] |
| Aichi Virus A (AiV-A) | Untreated wastewater | Duplex RT-LAMP | Not explicitly stated (Primary focus on LAMP development) | Developed for wastewater surveillance; performance compared internally between RT-qPCR and LAMP. | [101] |
| tet(M) Gene in Enterococci | Spiked human/animal urine | RT-LAMP with turbidimetry | LOD 100x more sensitive than conventional PCR | LOD: 0.001 pg/μL vs. 0.1 pg/μL for conventional PCR. 100% specificity demonstrated. | [102] |
Analysis of Discrepancies and Performance Drivers
The observed discrepancies in concordance, particularly the variable positive detection rates for SARS-CoV-2, can be attributed to several key factors central to a thesis investigation:
- Primer Design and Target Gene: The choice of target gene is a critical determinant of success. For SARS-CoV-2, primers targeting the Nsp3 (Set-4), S (Set-10, Set-11), and E (Set-13) genes demonstrated superior positive detection rates (75.9%-82.8%) compared to others [99]. This highlights the need for careful in silico analysis and empirical testing of multiple primer sets during assay development.
- Sample Type and Processing: LAMP's robustness to inhibitors is a significant advantage [6]. However, sample matrix can still impact performance. For instance, the tet(M) RT-LAMP assay was successfully validated in spiked human and animal urine, a complex biological matrix, demonstrating its practical applicability within a One Health framework [102]. The use of crude sample preparation methods, such as a simple boiling lysis for DNA extraction, further underscores the technique's utility in resource-limited settings [102].
- Limit of Detection (LOD): A core component of analytical sensitivity, the LAMP assay for pea root rot pathogens achieved an impressive LOD of 0.02 ng of gDNA and 10 spores per sample [6]. The tet(M) assay showed a 100-fold increase in sensitivity over conventional PCR [102]. These metrics are crucial for determining the clinical or environmental applicability of a LAMP assay.
Experimental Protocols for Comparative Analysis
To ensure the reproducibility of thesis findings, the following detailed protocols are provided for key experiments in the comparative evaluation of LAMP and RT-qPCR assays.
Protocol 1: Primer Efficiency Evaluation for Viral Detection
This protocol is adapted from the comparative evaluation of 19 SARS-CoV-2 RT-LAMP primer sets [99].
- Objective: To identify the most efficient primer set for a target pathogen.
- Materials:
- Purified RNA/DNA standards (e.g., in vitro transcribed RNA, synthetic genes, or genomic DNA).
- Candidate LAMP primer sets (typically 4-6 primers per set).
- WarmStart Bst 2.0 or 3.0 DNA Polymerase or equivalent RT-LAMP master mix.
- Real-time fluorometer or turbidimeter capable of isothermal amplification (e.g., BioRanger, Axxin T-series).
- Procedure:
- Reaction Setup: Prepare LAMP reactions according to the master mix manufacturer's instructions. Include 3000 copies of the RNA/DNA standard per reaction.
- Amplification: Run reactions at an isothermal temperature (typically 60-65°C) for 30-60 minutes with real-time monitoring.
- Data Analysis: Calculate the Time to threshold (Tt) for each reaction. Primer sets with faster amplification (lower Tt values) generally indicate higher efficiency and sensitivity.
- Selection: Select the top-performing primer sets (e.g., those with the lowest average Tt) for further validation with clinical samples.
Protocol 2: Clinical Sample Validation and Discrepancy Analysis
This protocol outlines the process for validating LAMP performance against RT-qPCR using clinical specimens [99] [100].
- Objective: To determine the clinical sensitivity, specificity, and concordance of the LAMP assay.
- Materials:
- A panel of well-characterized clinical samples (positive and negative for the target).
- RNA/DNA extraction kits (or materials for crude lysis, e.g., boiling).
- Optimized LAMP assay components.
- RT-qPCR assay components and thermocycler.
- Procedure:
- Sample Preparation: Extract nucleic acids from clinical samples using a standardized method. For robustness testing, include a subset processed via a rapid, crude method (e.g., 8-10 min boil followed by centrifugation) [102].
- Parallel Testing: Test all samples in parallel using the optimized LAMP assay and the reference RT-qPCR assay.
- Data Collection:
- For LAMP: Record Tt values and/or endpoint results (colorimetric change, fluorescence).
- For RT-qPCR: Record Ct values.
- Statistical Analysis:
- Calculate sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
- Determine the Cohen's Kappa (κ) statistic to measure agreement beyond chance. A κ > 0.8 indicates excellent agreement [100].
- Analyze discrepant results by retesting or using an alternative molecular method (e.g., digital PCR) to resolve the true status.
Protocol 3: Limit of Detection (LOD) Determination
This protocol describes how to determine the analytical sensitivity of the LAMP assay [6] [102].
- Objective: To establish the lowest concentration of the target that can be reliably detected by the LAMP assay.
- Materials:
- A calibrated standard with known copy number or concentration (e.g., synthetic gene, quantified amplicon, pathogen stock).
- Serial dilution buffers.
- Procedure:
- Sample Dilution: Prepare a logarithmic serial dilution (e.g., 10-fold) of the calibrated standard in a matrix that mimics the clinical sample (e.g., negative saliva, urine, or soil extract).
- Replication: Test each dilution level with a minimum of 8-12 replicates.
- Amplification: Run the LAMP assay on all replicates.
- Probit Analysis: Use statistical software (e.g., POD-LOD software) to determine the LoD95%, the concentration at which 95% of the replicates test positive.
Workflow and Signaling Pathway Diagrams
Experimental Workflow for LAMP vs. RT-qPCR Comparative Analysis
The following diagram illustrates the logical workflow for the direct comparison and discrepancy analysis between LAMP and RT-qPCR assays, as detailed in the protocols.
Figure 1: Experimental workflow for comparative assay analysis.
LAMP Reaction Signaling and Detection Pathways
LAMP amplification can be monitored through various signaling mechanisms. The following diagram outlines the primary detection pathways utilized in the assays discussed.
Figure 2: LAMP reaction signaling and detection pathways.
The Scientist's Toolkit: Research Reagent Solutions
The successful implementation of a LAMP assay requires specific reagents and tools. The following table details key components and their functions, as referenced in the studies.
Table 2: Essential Research Reagents for LAMP Assay Development
| Reagent / Tool | Function / Application | Specific Examples / Notes |
|---|---|---|
| Bst-like DNA Polymerase | Engineered DNA polymerase with strand-displacement activity essential for isothermal amplification. | WarmStart Bst 2.0 or 3.0, Bst-XT (NEB). WarmStart versions reduce non-specific amplification at room temperature [103]. |
| LAMP Primer Design Tool | Software to design the complex set of 4-6 primers required to target 6-8 distinct regions. | NEB LAMP Primer Design Tool, PrimerExplorer V5. Critical for ensuring assay specificity and efficiency [103] [102]. |
| Real-time Isothermal Instrument | Equipment for maintaining constant temperature and monitoring amplification in real-time. | BioRanger (Diagenetix), Axxin T8-ISO/T16-ISO. Enables quantification and Tt determination [104] [103]. |
| Colorimetric Master Mix | A pre-mixed solution containing buffer, dyes, and enzymes for visual endpoint detection. | WarmStart Colorimetric LAMP Master Mix (NEB). Allows result interpretation by visible color change (e.g., pink to yellow), ideal for point-of-care use [103] [104]. |
| Crude Lysis Reagents | Chemicals for rapid, equipment-free nucleic acid release, bypassing column-based extraction. | Double distilled water (ddH2O) for boiling method; Guanidinium chloride-containing buffers. Enhances speed and field-deployment capability [102] [104]. |
| Specificity Controls | Non-target DNA/RNA from related pathogens or host species to validate assay specificity. | Genomic DNA from cross-reactive organisms (e.g., other coronaviruses, enterococci species). Essential for confirming no off-target amplification [99] [102]. |
Limit of Detection (LoD) Benchmarking Across Viral Targets
Limit of Detection (LoD) benchmarking is a critical process in the validation of diagnostic assays, ensuring reliable, sensitive, and comparable results across different platforms and laboratories. For Loop-Mediated Isothermal Amplification (LAMP) assays, which are gaining prominence in rapid viral diagnostics due to their simplicity, speed, and portability, rigorous LoD determination is essential for confirming clinical utility [105] [64]. LAMP is an isothermal nucleic acid amplification technique that uses multiple primers for high specificity and efficiency. Its robustness makes it suitable for field-deployable diagnostics, but this also necessitates careful standardization to enable accurate comparison with established molecular methods like PCR [106] [6]. This application note provides a structured framework for LoD benchmarking of LAMP assays across diverse viral targets, supporting a broader thesis on advancing rapid viral diagnostic research.
Defining LoD and Key Performance Parameters
The Limit of Detection (LoD) is the lowest concentration of an analyte in a sample that can be reliably distinguished from a blank sample but does not necessarily be quantitated as an exact value [107]. It is a fundamental parameter characterizing an assay's sensitivity. Proper LoD determination requires understanding related metrics:
- Limit of Blank (LoB): The highest apparent analyte concentration expected from replicates of a blank sample. It is calculated as
LoB = mean_blank + 1.645(SD_blank)(assuming a one-sided 95% confidence interval for a Gaussian distribution) [108] [109]. - Limit of Quantitation (LoQ): The lowest concentration at which the analyte can be reliably detected and quantitated with acceptable precision and accuracy, typically defined by predetermined goals for bias and imprecision [108].
LoD is statistically derived using both the LoB and test replicates of a sample with low analyte concentration: LoD = LoB + 1.645(SD_low concentration sample) [108]. For visual or qualitative assays like many LAMP applications, LoD may be determined through logistic regression of detection rates at various concentrations, setting the LoD at a 99% detection probability [109].
LoD Benchmarking Data Across Diagnostic Technologies
Comparing LoD values across different assay types and viral targets highlights the relative performance of LAMP against other technologies and establishes expected sensitivity benchmarks.
Table 1: LoD Benchmarking Across Viral Detection Technologies
| Technology | Viral Target / Context | LoD | Key Findings / Context |
|---|---|---|---|
| LAMP | Hospital-acquired pneumonia pathogens (e.g., S. aureus, E. coli, P. aeruginosa) [64] | Determined for each bacterium in panel | The clinical sensitivity of the multiplex LAMP assay was 93.3% with a specificity of 92.0% when compared to culture. |
| LAMP | Pea root rot pathogens (A. euteiches, P. ultimum, F. solani) [6] | 0.02 ng of gDNA & 10 spores/sample | Demonstrated LAMP's high sensitivity and applicability in complex biological matrices like soil and root tissue. |
| Short-Read HTS (High-Throughput Sequencing) | Adventitious virus detection in biologics (EBV, FeLV, RSV, Reo1, PCV1) [110] | 104 GC/mL (detected by all labs); ≤102 GC/mL (achieved by some labs after optimization) | Highlights that optimization of the HTS workflow (sample processing, sequencing, bioinformatics) can significantly improve LoD. |
| RT-PCR (SARS-CoV-2 Assays) | SARS-CoV-2 diagnostic testing [111] | Variable across EUA assays | Cross-assay variation in LoD can impact clinical decision-making, especially for results near the assay's LoD. Viral loads < 106 copies/mL often not infectious. |
| HIV Viral Load Test (Clinical Standard) | HIV viral suppression monitoring [112] | <200 copies/mL defines viral suppression | This clinical threshold underscores the importance of ensuring diagnostic assays have LoDs sufficient to meet clinical and public health goals. |
Table 2: Comparison of LAMP with Other Molecular Detection Methods
| Method | Key Principle | Typical LoD | Advantages for Viral Diagnostics | Limitations |
|---|---|---|---|---|
| LAMP | Isothermal amplification with 4-6 primers [64] | Varies; can be single-digit copies or low ng DNA [6] [64] | Rapid (30-60 min), isothermal, portable, high specificity, robust [6] [64] | Multiplexing is challenging; primer design complexity [64] |
| PCR/qPCR | Thermal cycling for amplification | Varies; often very high sensitivity [106] | High sensitivity, quantitative, well-established, multiplexable [105] | Requires thermal cycler, longer run time, lab-based [6] |
| Rapid Diagnostic Tests (RDTs) | Immunoassay-based antigen detection | Generally higher than nucleic acid tests [106] | Very fast, low cost, no instrument needed | Lower sensitivity, especially low viral loads [106] |
| High-Throughput Sequencing (HTS) | Massively parallel sequencing of all nucleic acids in a sample [110] | ~104 GC/mL; can be lower with optimization [110] | Unbiased detection, novel virus discovery | High cost, complex data analysis, longer turnaround [110] |
Experimental Protocols for LoD Determination in LAMP Assays
Protocol 1: LoD Determination Using Quantitative Samples
This protocol is appropriate when a purified quantitative standard (e.g., synthetic DNA, quantified viral RNA) is available.
- Sample Preparation: Serially dilute the quantitative standard in the appropriate matrix (e.g., nuclease-free water, negative clinical matrix, TE buffer) across a range covering the expected LoD. A typical dilution series might be 10-fold, with 5-7 concentration levels [109] [107].
- Reaction Setup: For each dilution level, prepare a minimum of 20 replicates for a verification study, though establishment may require 60 replicates [108]. Include negative control replicates (no template) to confirm the LoB.
- LAMP Amplification: Perform the LAMP reaction under established optimal conditions (e.g., 60–65°C for 25–60 minutes) using a real-time detection system to monitor amplification or an end-point method with visual detection [6] [64].
- Data Analysis:
- For each dilution, calculate the proportion of positive replicates.
- Use probit or logistic regression analysis on the proportion of detected replicates versus the analyte concentration to determine the concentration at which 95% of the samples are detected [109] [107].
- Alternatively, if using the EP17 statistical method, calculate the LoB from negative controls and the LoD from the low-concentration sample as defined in Section 2 [108].
Protocol 2: LoD Determination for Viral Targets in a Clinical Matrix
This protocol validates the LAMP assay's sensitivity in a clinically relevant background, such as sputum or serum.
- Virus Stock and Spiking: Obtain a quantified virus stock. Create serial dilutions of the virus in the negative clinical matrix (e.g., BAL fluid, serum). The matrix should be confirmed negative for the target via a reference method [110] [64].
- Sample Processing/Nucleic Acid Extraction:
- For viscous samples (e.g., sputum, endotracheal aspirates), pre-treat with a liquefying solution containing DTT [64].
- Transfer an aliquot to a lysis buffer (e.g., RALF buffer) and heat at 99°C for 2 minutes for DNA extraction. Centrifuge briefly, and use the supernatant directly in the LAMP reaction [64].
- Optional: For potentially higher yields or more complex samples, use a commercial nucleic acid extraction kit.
- LAMP Reaction and Analysis: Follow steps 2-4 from Protocol 1. The use of a clinical matrix is crucial for determining the assay's true functional sensitivity in the presence of potential inhibitors.
Table 3: Key Research Reagent Solutions for LAMP Assay Development and LoD Benchmarking
| Reagent / Material | Function | Example / Specification |
|---|---|---|
| LAMP Primer Mix | Targets 6-8 distinct regions of the viral genome for highly specific amplification. | A set of 4-6 primers per target (F3, B3, FIP, BIP, Loop F, Loop B) designed using tools like NEB LAMP primer design tool [6]. |
| Isothermal DNA/RNA Polymerase | Enzymatic amplification at a constant temperature (60-65°C); some have reverse transcriptase activity for RNA viruses. | Bst 2.0 or 3.0 DNA Polymerase; warmStart RTx for reverse transcription LAMP [6]. |
| Lysis Buffer / DNA Extraction Kit | Releases and stabilizes nucleic acids from samples for amplification. | RALF buffer for rapid heat lysis [64]; DNeasy PowerSoil Pro Kit for complex samples [6]. |
| Quantified Viral Standard | Positive control for assay validation and creating standard curves for LoD determination. | Synthesized gBlocks, in vitro transcribed RNA, or commercially available quantified whole virus [110]. |
| Positive & Negative Controls | Validate each reaction's performance and identify contamination. | Must include a no-template control (NTC) and a positive control with known concentration near the LoD. |
Workflow and Decision Pathways for LoD Benchmarking
The following diagrams outline the experimental workflow for LoD determination and the decision pathway for interpreting benchmarked LoD values.
Diagram 1: LAMP Assay LoD Determination Workflow. This flowchart outlines the key steps for experimentally determining the Limit of Detection for a LAMP assay, from sample preparation to statistical analysis.
Diagram 2: LoD Benchmarking Decision Pathway. This decision tree guides the interpretation of a benchmarked LoD value against clinical and analytical requirements to determine assay suitability or the need for optimization.
Multi-site Validation Studies and Regulatory Considerations
Loop-mediated isothermal amplification (LAMP) has emerged as a critical nucleic acid amplification technique for rapid viral, bacterial, and parasitic pathogen detection. Its isothermal reaction conditions, rapid turnaround time, and tolerance to inhibitors make it particularly valuable for point-of-care testing in resource-limited settings [113] [114]. However, the transition from research development to clinical implementation requires rigorous multi-site validation studies and careful navigation of regulatory pathways. This application note provides a comprehensive framework for conducting multi-site validation of LAMP assays and addressing key regulatory considerations, with specific examples from successfully validated assays across diverse disease targets.
Multi-site Validation Framework
Core Validation Parameters and Performance Metrics
Multi-site validation studies for LAMP assays must establish analytical and clinical performance across multiple independent laboratories. The table below summarizes key performance metrics from published multi-site LAMP validation studies:
Table 1: Performance Metrics from Multi-site LAMP Validation Studies
| Pathogen | Sample Type | Sensitivity (%) | Specificity (%) | Sample Size | Reference Method | Study Sites |
|---|---|---|---|---|---|---|
| SARS-CoV-2 [115] | Mid-turbinate swab | 88.2 | 100 | 1,378 participants | RT-PCR | 4 |
| SARS-CoV-2 [115] | Nasopharyngeal | 65.4 | 97.6 | 1,378 participants | RT-PCR | 4 |
| Salmonella [116] | Dry dog food | Comparable to BAM method | Comparable to BAM method | 24 samples per collaborator | FDA BAM culture | 7 |
| Mycobacterium tuberculosis [117] | Sputum | 89.4 | 94.1 | 350 | MGIT culture | 1 |
| Escherichia coli [118] | Urine | 100 | 92.7 | 440 | Culture | 1 |
These studies demonstrate that performance varies significantly based on sampling method, sample processing, and study design. The SARS-CoV-2 study highlighted substantial inter-site variability, with sensitivity ranging from 63.8% to 89.1% across different locations, emphasizing the importance of multi-site validation [115].
Implementation Considerations Across Settings
Successful deployment of LAMP technology requires attention to operational factors beyond pure analytical performance:
- Training Requirements: Laboratory technicians with no prior LAMP experience demonstrated significant improvement in knowledge and technical skills after structured training, with self-assessed competency increasing from a median score of 1/10 to 10/10 after training and follow-up [114].
- Sample Processing: Simplified, extraction-free protocols have been successfully implemented for HPV detection, reducing processing time and complexity while maintaining performance [119].
- Platform Selection: Both commercial systems (Genie II/III, Axxin T8-ISO) and laboratory-developed platforms have been successfully validated across different settings [116] [119].
Experimental Protocols for Multi-site Validation
Standardized Multi-laboratory Validation Protocol
The following protocol adapts methodologies from successfully validated LAMP assays for Salmonella detection in animal food [116]:
Sample Preparation and Inoculation
- Obtain matrix-free samples (e.g., dry dog food) and confirm negative status for target pathogen
- Prepare separate sample sets: uninoculated controls, low-level inoculations (0.65 MPN/25 g), and high-level inoculations (3.01 MPN/25 g)
- Blind-code all samples and distribute to participating laboratories
- Ensure synchronized initiation of testing across all sites
DNA Extraction and Amplification
- For each test portion, add 25 g sample to 225 mL pre-enrichment broth (buffered peptone water)
- Incubate at 37°C for 20-24 hours
- Extract DNA using magnetic bead-based purification systems
- Prepare LAMP master mix containing:
- 1.6 µM each of FIP and BIP primers
- 0.4 µM each of F3 and B3 outer primers
- 0.2 µM each of LF and LB loop primers
- 1X WarmStart LAMP Master Mix
- 2 µM intercalating dye (SYTO 16, SYBR Safe, or equivalent)
- Aliquot 20 µL master mix per reaction
- Add 5 µL DNA template (1.5 ng/µL concentration)
- Run amplification at 63-65°C for 40-60 minutes in real-time detection system
Data Analysis and Concordance Assessment
- Establish threshold values for positive amplification based on positive and negative controls
- Confirm all LAMP-positive samples by reference method culture
- Test all reference method-positive samples by LAMP
- Calculate probability of detection (POD) and statistical differences using random intercept logistic regression models
Integrated LAMP-CRISPR/Cas12a Protocol for Enhanced Specificity
For targets requiring ultra-specific detection, such as respiratory pathogens in pediatric populations, LAMP can be combined with CRISPR-Cas12a technology [120]:
Sample Processing
- Collect clinical samples (nasopharyngeal swabs, bronchoalveolar lavage fluid) in appropriate transport media
- Extract nucleic acids using magnetic bead-based kits (e.g., JIFA 502-B type)
- Add 20 μL Proteinase K, 10 μL magnetic beads, and 500 μL buffer to 200 μL sample
- Incubate at room temperature for 10 minutes
- Wash three times with wash buffer
- Elute in 50-100 μL RNase-free water at 70°C for 2.5 minutes
LAMP-CRISPR/Cas12a Reaction
- Perform LAMP amplification with target-specific primers (e.g., cpsA for S. pneumoniae, p1 for M. pneumoniae)
- Program: 65°C for 40 minutes
- Prepare CRISPR-Cas12a detection mix:
- 50 nM Cas12a enzyme
- 50 nM specific crRNA
- 500 nM fluorescent reporter (e.g., FAM-TTATTATT-BHQ1)
- 1X NEBuffer 2.1
- Combine 5 μL LAMP product with 15 μL CRISPR detection mix
- Incubate at 37°C for 10 minutes
- Measure fluorescence with plate reader or lateral flow detection
Table 2: Research Reagent Solutions for LAMP Validation
| Reagent Category | Specific Examples | Function | Implementation Considerations |
|---|---|---|---|
| Primer Sets | F3, B3, FIP, BIP, LF, LB [117] | Target-specific amplification | Design using LAMP designer software; optimize concentrations (1.6 µM FIP/BIP, 0.4 µM F3/B3, 0.2 µM LF/LB) |
| Enzymes | Bst DNA polymerase (WarmStart LAMP Master Mix) [117] | Strand displacement DNA synthesis | Select isothermal polymerase with high strand displacement activity |
| Detection Chemistries | SYTO 16, SYBR Safe, DARQ LAMP [117] [119] | Real-time amplicon detection | DARQ LAMP provides specific detection; intercalating dyes offer simplicity |
| Extraction Kits | Magnetic bead-based kits (MoBio, JIFA 502-B) [121] [120] | Nucleic acid purification | Magnetic protocols reduce hands-on time; some applications permit extraction-free direct detection [119] |
| Controls | Recombinant plasmid (pL-mpt64) [117] | Assay performance verification | Clone target gene into plasmid vector for quantitative positive control |
Regulatory Considerations
FDA Emergency Use Authorization Pathways
The U.S. Food and Drug Administration (FDA) has established specific pathways for molecular diagnostic tests, with numerous LAMP-based assays receiving Emergency Use Authorization (EUA) during the COVID-19 pandemic [122]. Key regulatory considerations include:
Target Selection and Verification
- Multiple target designs are preferred over single targets due to reduced vulnerability to viral mutations [122]
- Analytical sensitivity must be established through limit of detection studies with serial dilutions of quantified standards
- Analytical specificity should be verified against near-neighbor organisms and human genomic DNA
Clinical Performance Data
- Sites should include diverse geographic representation where applicable
- Predominantly symptomatic populations should be included with stratification by duration of symptoms
- Asymptomatic testing claims require specific validation with appropriate populations
Quality Control Requirements
- Internal controls must be incorporated to monitor extraction and amplification efficiency
- External controls should be included in each run at multiple concentrations near the assay limit of detection
The following diagram illustrates the complete workflow from multi-site validation to regulatory authorization for LAMP assays:
Technology Transfer and Training Requirements
Successful multi-site implementation requires comprehensive technology transfer programs:
- Structured Training Curriculum: Combine theoretical foundations with hands-on practical sessions using actual instrumentation and reagents [114]
- Competency Assessment: Evaluate technical staff through pre- and post-training questionnaires and direct observation during follow-up visits
- Ongoing Support: Schedule follow-up visits at 1-month and 6-month intervals to reinforce skills and troubleshoot challenges
Multi-site validation represents a critical step in the translation of LAMP assays from research tools to clinically implemented diagnostics. Successful validation requires careful attention to study design, standardized protocols, comprehensive training, and understanding of regulatory requirements. The frameworks and protocols presented herein, drawn from successfully validated LAMP assays across diverse applications, provide a roadmap for researchers and developers seeking to advance LAMP technologies through the validation and regulatory pathway. As LAMP technology continues to evolve with innovations such as extraction-free protocols, CRISPR integration, and portable detection platforms, robust multi-site validation will remain essential for establishing clinical utility and gaining regulatory approval.
Performance in Asymptomatic and Low Viral Load Scenarios
Reverse Transcription Loop-Mediated Isothermal Amplification (RT-LAMP) has emerged as a pivotal molecular technology for rapid viral diagnostics, particularly during the COVID-19 pandemic. Its value proposition centers on simplicity, speed, and cost-effectiveness compared to the gold standard RT-PCR. However, a thorough evaluation of its performance in challenging diagnostic scenarios—specifically asymptomatic infections and low viral load cases—is essential for determining its appropriate applications within research and clinical settings. This application note synthesizes current evidence to guide researchers, scientists, and drug development professionals on the capabilities and limitations of RT-LAMP under these critical conditions, providing detailed protocols for implementation.
Performance Data Analysis
The diagnostic sensitivity of RT-LAMP is intrinsically linked to the viral load present in the sample. The following table summarizes key performance metrics from recent studies, with viral load often approximated via RT-PCR Cycle Threshold (Ct) values.
Table 1: Performance of RT-LAMP in Asymptomatic and Low Viral Load Scenarios
| Study & Sample Type | Population/Sensitivity Context | Overall Sensitivity (%) | Sensitivity (Ct ≤ 25-33) | Sensitivity (Ct > 33-35) | Specificity (%) |
|---|---|---|---|---|---|
| Direct Saliva RT-LAMP (with RapiLyze) [123] | Asymptomatic & Symptomatic | 84.6 | 99.0 (Ct≤33) | N/R | ~100 |
| Direct Saliva RT-LAMP [124] | Asymptomatic | 40.9 - 45.0 | N/R | N/R | ~100 |
| Colorimetric RT-LAMP (Prospective Field Trial) [96] | Suspect Cases & Asymptomatic | 87.0 | 97.0 (Ct<35) | 60.0 (Ct≥35) | 98.0 |
| RNA RT-LAMP (Swabs) [123] | Asymptomatic & Symptomatic | 96.1 | N/R | N/R | ~100 |
| RT-LAMP (Meta-Analysis) [125] | Mixed | 79.0 (Overall) | N/R | N/R | 97.0 |
| RT-LAMP (with RNA Extraction) [125] | Mixed | 88.0 | N/R | N/R | N/R |
| RT-LAMP (without RNA Extraction) [125] | Mixed | 50.0 | N/R | N/R | N/R |
N/R = Not Reported
Data synthesis reveals a consistent trend: RT-LAMP exhibits high sensitivity (often >95%) in samples with high viral loads (typically corresponding to RT-PCR Ct values <25-35) [123] [96]. This makes it a reliable tool for identifying highly infectious individuals. However, its performance drops significantly in samples with low viral loads (Ct >33-35), with sensitivity reported as 60% [96] or lower. This limitation is particularly relevant for asymptomatic screening, where viral loads can be highly variable. One study focusing on asymptomatic individuals reported a sensitivity as low as 40.9%-45% for a Direct RT-LAMP format without RNA extraction [124]. Crucially, the inclusion of an RNA extraction step prior to RT-LAMP consistently improves sensitivity, as evidenced by the meta-analysis showing 88% sensitivity with extraction versus 50% without [125].
Experimental Protocols
To achieve optimal performance in asymptomatic and low viral load settings, robust and sensitive protocols are essential. The following sections detail two optimized methodologies.
Protocol 1: High-Sensitivity RT-LAMP with RNA Extraction
This protocol is recommended for scenarios where maximizing detection sensitivity is the highest priority, such as in research settings or for confirming low-positive results [125] [123].
- Sample Collection: Collect nasopharyngeal/oropharyngeal swabs and place in Viral Transport Medium (VTM). For saliva, instruct participants to self-collect drooled saliva in a sterile container, abstaining from eating, drinking, or oral hygiene for 30 minutes prior [126] [123].
- RNA Extraction:
- Use approved commercial kits such as the Maxwell RSC Viral Total Nucleic Acid Purification Kit (Promega), MagMAX CORE Nucleic Acid Purification Kit (Thermo Fisher), or QIAsymphony Virus/Bacteria Mini Kit (Qiagen) [123].
- Follow manufacturer instructions. For example, using the Maxwell RSC system, mix 200μL of sample with 223μL of lysis buffer, incubate for 10 minutes at room temperature and 10 minutes at 56°C for inactivation, then complete automated extraction, eluting in 50μL of nuclease-free water [123].
- RT-LAMP Reaction:
- Primers: Utilize primer sets targeting SARS-CoV-2 genes (e.g., N, E, Orf1ab). The E-ID1 primer set (5 primers) has demonstrated reduced false-positive rates [13].
- Master Mix: Assemble a reaction containing:
- Additives: To enhance speed and efficiency, include 40mM Guanidine Hydrochloride (GuHCl) [13].
- Amplification: Incubate reactions at 65°C for 20-40 minutes.
- Detection:
Diagram 1: High-Sensitivity RT-LAMP Workflow
Protocol 2: Direct Saliva RT-LAMP for Rapid Screening
This streamlined protocol is suitable for high-throughput, frequent screening of asymptomatic populations where speed and resource conservation are prioritized over absolute maximum sensitivity [123].
- Sample Collection and Preparation:
- Collect drooled saliva as described in Protocol 1.
- Mix 50μL of saliva with 50μL of RapiLyse buffer (OptiGene) in a reaction tube [123].
- Heat the mixture at 98°C for 2 minutes for viral inactivation and RNA release [123].
- Centrifuge briefly to pellet debris. The supernatant is used directly in the RT-LAMP reaction.
- RT-LAMP Reaction:
- Detection: As described in Protocol 1. Due to the lack of purification, careful interpretation of faint color changes is advised.
The Scientist's Toolkit
Successful implementation of these protocols relies on key reagents and components. The following table outlines essential solutions and their functions.
Table 2: Key Research Reagent Solutions for RT-LAMP
| Reagent / Component | Function / Role | Examples & Notes |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase; engine of the isothermal amplification. | Bst 2.0 (NEB) offers robust performance. Bst 3.0 (NEB) has integrated RT activity for single-enzyme reactions [13]. |
| Primer Sets | Specifically bind target sequences to initiate amplification. | 5-primer sets (e.g., E-ID1) can reduce false positives. 6-primer sets (e.g., targeting N, S genes) are common [13]. |
| Colorimetric Dye | Enables visual readout of amplification by pH change. | Phenol Red. A color shift from pink (basic) to yellow (acidic) indicates a positive reaction [127]. |
| Sample Lysis Buffer | Inactivates virus and releases RNA for direct testing. | RapiLyse (OptiGene) simplifies saliva processing. Chelex 100 Resin is also used [123]. |
| Reaction Enhancer | Improves amplification speed, efficiency, and robustness. | Guanidine Hydrochloride (GuHCl) can reduce detection time and enhance signal [13]. |
| Positive Control | Validates the entire testing process from extraction to amplification. | Inactivated SARS-CoV-2 virus or synthetic RNA controls (e.g., Twist synthetic RNA) [13] [123]. |
The body of evidence indicates that RT-LAMP is a highly effective diagnostic tool for identifying individuals with medium to high viral loads, a group considered highly infectious. Its speed, cost-effectiveness, and simplicity make it exceptionally suitable for rapid screening programs and deployment in resource-limited settings [128] [96].
However, for applications involving asymptomatic individuals or cases with low viral loads, protocol selection is critical. The significant reduction in sensitivity for low viral load samples is the primary limitation [124] [96]. To mitigate this, incorporating an RNA extraction step is the most impactful modification to enhance sensitivity [125] [126]. Furthermore, the use of optimized primer sets (e.g., 5-primer systems) and reaction enhancers like GuHCl can improve reliability and reduce false positives [13].
In conclusion, while RT-LAMP may not replace RT-PCR in scenarios requiring the utmost sensitivity, it represents a powerful alternative within a stratified testing strategy. Researchers and clinicians can confidently employ optimized RT-LAMP protocols for rapid identification of infectious cases, but should confirm negative results with RT-PCR when there is a high clinical suspicion of infection, particularly in asymptomatic and low viral load scenarios.
Cost-Effectiveness Analysis and Operational Efficiency Metrics
Loop-mediated isothermal amplification (LAMP) has emerged as a transformative nucleic acid amplification technique, offering a compelling alternative to conventional polymerase chain reaction (PCR) in diagnostic and research applications. Its operational simplicity, high sensitivity, and ability to function under isothermal conditions make it particularly valuable for resource-limited settings and point-of-care testing [59]. This application note provides a comprehensive analysis of the cost-effectiveness and operational efficiency metrics of LAMP assays, contextualized within viral diagnostics research. We present synthesized quantitative data, detailed experimental protocols, and analytical frameworks to enable researchers to evaluate and implement LAMP technology effectively across diverse applications from clinical diagnostics to agricultural pathogen detection.
The growing adoption of LAMP is reflected in market projections, with the global LAMP market expected to grow from USD 115.7 million in 2025 to USD 184.8 million by 2035, representing a compound annual growth rate (CAGR) of 4.9% [129]. This growth is driven by increasing demand for rapid, accurate, and affordable diagnostic solutions, particularly in infectious disease detection. Our analysis demonstrates that LAMP's economic advantage stems from multiple factors: minimal equipment requirements, reduced energy consumption, rapid turnaround times, and compatibility with simplified detection methods that eliminate the need for expensive instrumentation [59] [80].
Comparative Cost-Effectiveness Analysis
Economic Evaluation of LAMP in Clinical Diagnostics
Numerous studies have demonstrated the cost-effectiveness of LAMP compared to traditional diagnostic methods. A recent systematic review of economic evaluations for pulmonary tuberculosis diagnosis found that both Xpert MTB/RIF and TB-LAMP were either cost-saving or highly cost-effective compared to conventional smear microscopy and culture methods [130]. The analysis, which standardized costs to 2025 US dollars, revealed that molecular tests including LAMP provide significant value despite higher initial cartridge costs, due to their superior sensitivity and faster time-to-diagnosis which reduces transmission and improves patient outcomes.
Table 1: Cost-Effectiveness Comparison of Diagnostic Methods for Pulmonary Tuberculosis
| Diagnostic Method | Sensitivity Range | Specificity Range | Cost per Test (USD) | Time to Result | Incremental Cost-Effectiveness Ratio |
|---|---|---|---|---|---|
| Smear Microscopy | 40-50% | >95% | $3-5 | Hours | Reference |
| Culture | >95% | >99% | $15-30 | 2-6 weeks | $500-1,000 per DALY averted |
| Xpert MTB/RIF | ≥85% | >95% | $9.98 | <2 hours | Cost-saving to $210 per DALY averted |
| TB-LAMP | ≥85% | >95% | $7-12 | <1 hour | Cost-saving to $180 per DALY averted |
In the context of antimicrobial resistance detection, a distance-based paper device (dPAD) LAMP assay for ESBL-producing E. coli demonstrated significant cost advantages, providing results within 42 minutes at a fraction of the cost of quantitative PCR (qPCR) [131]. The assay achieved sensitivity and specificity of 98.9% and 96.5% respectively, while utilizing inexpensive paper-based detection that eliminated the need for expensive fluorometers or spectrophotometers.
Operational Efficiency Metrics Across Applications
LAMP technology demonstrates remarkable operational efficiency across diverse applications, from clinical diagnostics to agricultural testing. The following table synthesizes key performance metrics from recent implementations:
Table 2: Operational Efficiency Metrics of LAMP Assays Across Applications
| Application | Target | Time to Result | Limit of Detection | Specificity | Cost per Sample |
|---|---|---|---|---|---|
| UTI Diagnosis [131] | ESBL E. coli | 42 minutes | 10 CFU/mL | 96.5% | <$5 (excluding labor) |
| ABO Genotyping [132] | Blood group SNPs | <60 minutes | 10 copies | 97.4-100% | $8-12 |
| Plant Pathogen Detection [6] | Pea root rot pathogens | 60 minutes | 0.02 ng gDNA | High specificity | <$10 |
| Sunflower Mildew Detection [23] | Plasmopara halstedii | 35-45 minutes | 0.5 pg/μL | High specificity | <$8 |
| COVID-19 Detection [80] | SARS-CoV-2 RNA | 30-60 minutes | 25-50 copies/μL | 100% | $2.9-6 |
The operational efficiency of LAMP assays is further enhanced by their tolerance to inhibitors commonly found in clinical and environmental samples, reducing the need for extensive nucleic acid purification [10]. This robustness makes LAMP particularly suitable for direct detection from crude samples, streamlining workflow and reducing processing time and costs.
Experimental Protocols
Protocol 1: Lyophilized RT-LAMP for RNA Virus Detection
This protocol adapts the open-source RT-LAMP method for detection of RNA viruses, utilizing non-proprietary enzymes to reduce costs while maintaining high sensitivity and specificity [80].
Reagent Preparation
Enzyme Mix Formulation:
- HIV-1 reverse transcriptase (in-house purified or commercial): 1.2 μL (25 U/μL)
- Bst LF DNA polymerase (in-house purified or commercial): 1.0 μL (8,000 U/mL)
- Thermolabile UDG (optional): 0.2 μL (5 U/μL)
- Nuclease-free water: to final volume
Lyophilized Reaction Mix (per reaction):
- Trehalose: 1.5 μL (1M stock)
- Primers (FIP/BIP: 1.6 μM each; F3/B3: 0.2 μM each; LF/LB: 0.4 μM each)
- dNTPs: 1.4 mM
- Betaine: 0.8 M
- MgSO₄: 6 mM
- Neutral Red dye: 0.2 mM
- Aliquot 12.5 μL per tube and lyophilize for 4 hours
Assay Procedure
- Sample Preparation: Mix 5 μL of raw sample (nasopharyngeal swab in transport media) with 5 μL of sample preparation buffer (50 mM KOH, 0.1% Triton X-100).
- Reaction Setup: Add 15 μL of nuclease-free water to lyophilized reaction pellet, followed by 5 μL of treated sample.
- Amplification: Incubate at 65°C for 45 minutes using a heating block or water bath.
- Detection: Visualize color change from orange to pink/red for positive samples. For quantitative analysis, use a portable fluorometer.
Performance Validation
- Analytical Sensitivity: Determine limit of detection using serial dilutions of synthetic RNA (typically 10-50 copies/μL).
- Specificity Testing: Validate against a panel of related pathogens to ensure no cross-reactivity.
- Storage Stability: Lyophilized reagents maintain performance for ≥3 months at ambient temperature.
Protocol 2: Distance-Based Paper Device (dPAD) LAMP for Bacterial Load Quantification
This protocol describes a semi-quantitative LAMP approach using a paper-based device for detection and approximate quantification of bacterial pathogens, with application in antimicrobial resistance monitoring [131].
dPAD Fabrication
- Material Preparation:
- Cut chromatography paper to 30 × 60 mm dimensions
- Create hydrophobic barriers using wax printing (120°C for 1.5 minutes)
- Apply 2% polyethyleneimine (PEI) solution to detection zone
- Dry at 37°C for 30 minutes
- Seal with transparent adhesive tape
LAMP Reaction Setup
Reaction Mix (25 μL total volume):
- 1× Isothermal Amplification Buffer
- 6 mM MgSO₄
- 1.4 mM dNTPs
- 1.6 μM FIP/BIP primers
- 0.2 μM F3/B3 primers
- 0.4 μM LF/LB primers
- Fluorescent dye (SYBR Green or equivalent): 0.5 μL
- Bst 2.0 Warm Start DNA Polymerase: 8 U
- Template DNA: 2 μL
Amplification Conditions:
- Incubate at 63°C for 30 minutes
- Heat inactivation at 80°C for 5 minutes
Detection and Quantification
- Apply 6 μL of LAMP reaction mixture to sample zone of dPAD
- Allow capillary flow for 5-10 minutes
- Measure migratory distance of fluorescent front under UV light
- Correlate distance to bacterial load using standard curve (10²-10⁶ CFU/mL)
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of LAMP assays requires careful selection of reagents and materials. The following table outlines key components and their functions in LAMP-based research applications:
Table 3: Essential Research Reagents for LAMP Assay Development
| Reagent/Material | Function | Recommended Specifications | Alternative Formulations |
|---|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase for isothermal amplification | Bst LF or Bst 2.0 (8,000 U/mL) | In-house purified Bst polymerase [80] |
| Reverse Transcriptase | RNA template conversion to cDNA (for RT-LAMP) | HIV-1 RT or WarmStart RTx | M-MuLV RT for lower temperature applications |
| Primer Sets | Target-specific amplification (6-8 regions) | HPLC-purified, 0.2-1.6 μM final concentration | Modified primers for detection (biotin, FAM) [10] |
| Colorimetric Indicators | Visual detection of amplification | Neutral Red (0.2 mM), Hydroxynaphthol Blue (120 μM) | Phenol Red, SYBR Green [23] |
| Lyoprotectants | Stabilization of lyophilized reagents | Trehalose (0.2-0.5 M) | Sucrose, mannitol [80] |
| Metal Cofactors | Enzyme activity and reaction efficiency | MgSO₄ (6-8 mM) | MgCl₂ with concentration optimization |
| Betaine | Secondary structure destabilization | 0.6-1.0 M | DMSO for GC-rich targets |
Technological Integration and Workflow Optimization
The integration of LAMP with emerging technologies significantly enhances its operational efficiency and application scope. Microfluidic systems represent a particularly promising direction, with this segment expected to account for 63.8% of the LAMP market share in 2025 [129]. These systems enable the automation of sample preparation, amplification, and detection within compact devices, reducing manual handling and improving reproducibility.
Recent innovations in detection methodologies have further expanded LAMP's utility. Molecular beacon (MB)-LAMP and nucleic acid lateral flow (NALF) detection provide enhanced specificity through sequence-specific probing, reducing false positives [10]. These approaches maintain the operational simplicity of LAMP while improving diagnostic accuracy, making them particularly valuable for clinical applications where specificity is critical.
For agricultural applications, such as detection of pea root rot pathogens, LAMP protocols have been optimized for direct use with soil and plant tissue samples, eliminating the need for DNA purification [6]. This streamlined workflow reduces processing time from days to under 60 minutes while maintaining sensitivity equivalent to qPCR, demonstrating the remarkable operational efficiency achievable through method optimization.
LAMP technology represents a paradigm shift in molecular diagnostics, offering compelling advantages in cost-effectiveness and operational efficiency compared to traditional amplification methods. The quantitative data and protocols presented in this application note demonstrate that well-optimized LAMP assays can achieve sensitivity and specificity comparable to PCR while significantly reducing equipment requirements, processing time, and per-test costs. The ongoing development of lyophilized reagents, portable detection platforms, and simplified sample preparation methods continues to expand LAMP's applicability across diverse settings from advanced laboratories to resource-limited field environments.
For researchers implementing LAMP assays, careful attention to primer design, reaction optimization, and appropriate detection methodology selection is crucial for achieving optimal performance. The integration of LAMP with emerging technologies such as microfluidics, paper-based detection, and smartphone-based readout systems promises to further enhance its accessibility and operational efficiency, positioning LAMP as a cornerstone technology for the next generation of molecular diagnostics.
Conclusion
LAMP technology represents a paradigm shift in viral diagnostics, offering rapid, sensitive, and equipment-independent testing capabilities crucial for both laboratory and point-of-care applications. The integration of advanced detection formats, robust primer design strategies, and optimized protocols has enabled reliable detection of diverse pathogens including SARS-CoV-2 variants, MPXV clades, and Ebola virus with performance comparable to gold-standard methods. Future directions should focus on developing multiplexed panels for syndromic testing, creating stable lyophilized reagents for distribution, integrating with microfluidic and smartphone-based platforms, and expanding applications to emerging pathogens and antimicrobial resistance detection. As the technology continues to evolve, LAMP assays hold significant potential to enhance global disease surveillance capabilities and accelerate therapeutic development through rapid diagnostic implementation.
References
- 1. Advancements and applications of loop-mediated isothermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11292733/]
- 2. Loop Mediated Isothermal Amplification: Principles and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7238132/]
- 3. Loop-mediated isothermal amplification [https://en.wikipedia.org/wiki/Loop-mediated_isothermal_amplification]
- 4. Getting started with loop-mediated isothermal amplification ... [https://www.neb.com/en-us/nebinspired-blog/getting-started-with-loop-mediated-isothermal-amplification?srsltid=AfmBOopjGoFus_j8450Fd4iBkRJKqBt4PI9UoKEQ_OPC-rTtk6r0e8y1]
- 5. Recent advances in loop-mediated isothermal ... [https://www.sciencedirect.com/science/article/pii/S2666517422000177]
- 6. Loop mediated isothermal amplification (LAMP) as a rapid ... [https://www.nature.com/articles/s41598-025-18738-9]
- 7. Visualization methods for loop mediated isothermal ... [https://pubmed.ncbi.nlm.nih.gov/39895350/]
- 8. Colorimetric LAMP/RT-LAMP Protocol [https://www.protocols.io/view/colorimetric-lamp-rt-lamp-protocol-ci5kug4w]
- 9. Loop mediated isothermal Amplification & Lateral Flow [https://www.milenia-biotec.com/en/method/loop-mediated-isothermal-amplification/]
- 10. Development of loop-mediated isothermal amplification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12442385/]
- 11. Loop-Mediated Isothermal Amplification [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/isothermal-amplification/loop-mediated-isothermal-amplification-lamp?srsltid=AfmBOooQFMOf9ANyD_k-ZP5r8OvI2Q4K6mug3kveO7mLKbMt8sq2Bh9F]
- 12. Isothermal Nucleic Acid Amplification Techniques (INAAT) [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/isothermal-nucleic-acid-amplification.html]
- 13. Development of loop-mediated isothermal amplification ... [https://www.nature.com/articles/s41598-023-31760-z]
- 14. Loop‐mediated isothermal amplification (LAMP) [https://pmc.ncbi.nlm.nih.gov/articles/PMC7167136/]
- 15. Engineering and enhancement of Bst DNA polymerase ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025092487]
- 16. Charge Engineering Improves the Performance of Bst DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9514385/]
- 17. Lamp Assays - TekTalk Newsletter [https://www.agilent.com/about/tektalk/en/newsletter-lamp-assays.html?srsltid=AfmBOopSe71BmaWkV3aN1drixlRljuoCs1eeEUEOu2olix7F7BUFuMUe]
- 18. Multiplexing LAMP Assays: A Methodological Review and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11203869/]
- 19. Multiple Endonuclease Restriction Real-Time Loop ... [https://www.sciencedirect.com/science/article/pii/S1525157815000720]
- 20. Development of loop-mediated isothermal amplification assay ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-14-10]
- 21. Loop-mediated isothermal amplification of DNA - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC102748/]
- 22. Loop-Mediated Isothermal Amplification (LAMP): The Better ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8393631/]
- 23. Optimization of loop-mediated isothermal amplification ... [https://www.nature.com/articles/s41598-024-72228-y]
- 24. Loop-mediated isothermal amplification (LAMP) assay for ... [https://www.nature.com/articles/s41598-022-22023-4]
- 25. A colorimetric RT-LAMP assay and LAMP-sequencing for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7574920/]
- 26. Optimization of loop-mediated isothermal amplification (LAMP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8757654/]
- 27. Comparison of diagnostic performance of RT-qPCR, RT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10170900/]
- 28. Comparison of RT-LAMP and RT-qPCR assays for ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093423001969]
- 29. Advancements and applications of loop-mediated ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1406632/full]
- 30. Evaluation and comparison of one-step real-time PCR and ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-09574-9]
- 31. Rapid isothermal amplification and portable detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7502724/]
- 32. Diagnostic accuracy of LAMP versus PCR over the course of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8056478/]
- 33. A Simple, Affordable, Rapid, Stabilized, Colorimetric, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8000859/]
- 34. Advancements in LAMP -Based Diagnostics: Emerging Techniques and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11546353/]
- 35. Frontiers | A rapid and visual dual LAMP -LFD assay for on-site... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1678396/full]
- 36. Development and validation of a multi-platform LAMP system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12619727/]
- 37. Multiplexed detection of respiratory viral by isothermal... pathogens [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00509d]
- 38. Development and Validation of LAMP Assays for ... [https://www.mdpi.com/2079-6374/15/1/23]
- 39. A paper-based loop-mediated isothermal amplification assay ... [https://pubmed.ncbi.nlm.nih.gov/40204842/]
- 40. A paper-based loop-mediated isothermal amplification ... [https://pubs.rsc.org/en/content/articlelanding/2025/ay/d5ay00749f]
- 41. Loop-Mediated Isothermal Amplification (LAMP) [https://www.thermofisher.com/us/en/home/life-science/pcr/isothermal-nucleic-acid-amplification/loop-mediated-isothermal-amplification.html]
- 42. Clinical Validation of a Colorimetric Loop-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10093461/]
- 43. An alternative real-time fluorescence reverse transcription ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204207/]
- 44. Enhancing Rapid Detection of COVID-19: Optimization ... [https://brieflands.com/journals/jjm/articles/160288]
- 45. Standardization and performance of SARS-CoV-2 RT- ... [https://pubmed.ncbi.nlm.nih.gov/40956555/]
- 46. A Multiplex and Colorimetric Reverse Transcription Loop ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2021.653616/full]
- 47. Detecting SARS-CoV-2 at point of care: preliminary data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7574392/]
- 48. Critical aspects of droplet digital reverse transcription loop ... [https://www.nature.com/articles/s41378-025-00982-8]
- 49. Rapid, multiplex detection of SARS-CoV-2 using ... [https://www.nature.com/articles/s41598-022-27133-7]
- 50. Modeling the effectiveness of RT-PCR, RT-LAMP, and antigen ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-025-11793-7]
- 51. Comparative analysis of Mpox clades - BMC Infectious Diseases [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-025-11784-8]
- 52. Clade I or clade II? Targeting essential viral genes to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12354367/]
- 53. Development of a Loop-Mediated Isothermal Amplification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9603857/]
- 54. Extraction-free LAMP assays for generic detection of Old ... [https://www.nature.com/articles/s41598-023-48391-z]
- 55. Rapid and highly sensitive colorimetric LAMP assay ... [https://www.sciencedirect.com/science/article/abs/pii/S000326702400521X]
- 56. Detection of Monkeypox Virus Clade Ib DNA in Wastewater ... [https://wwwnc.cdc.gov/eid/article/31/10/25-0270_article]
- 57. Development of a multiplex real-time PCR assay for the ... [https://www.sciencedirect.com/science/article/pii/S0166093424000818]
- 58. Loop-Mediated Isothermal Amplification [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/isothermal-amplification/loop-mediated-isothermal-amplification-lamp?srsltid=AfmBOootobZx7Fh8gXdpnHZg9F1mtUN_ObUpAJcDRXhFI18KGGGADS4D]
- 59. Visualization methods for loop mediated isothermal ... [https://pubs.rsc.org/en/content/articlehtml/2025/an/d4an01287a]
- 60. Evolution of the Probe-Based Loop-Mediated Isothermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10177487/]
- 61. Recent advances in loop-mediated isothermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9325740/]
- 62. A new approach for the detection of genetic alterations ... [https://www.nature.com/articles/s41598-025-93086-2]
- 63. Data treatment methods for real-time colorimetric loop ... [https://www.nature.com/articles/s41598-023-40737-x]
- 64. Assessment of a rapid diagnostic test based on loop ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1609666/full]
- 65. Comparison of DPO-based PCR, RT-PCR and LAMP for ... [https://www.sciencedirect.com/science/article/abs/pii/S0044848625004533]
- 66. Development of a large-scale rapid LAMP diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11634539/]
- 67. Establishment of Sample-to-Answer Loop-Mediated ... [https://www.mdpi.com/2079-6374/14/12/609]
- 68. A semi-automated, isolation-free, high-throughput SARS ... [https://www.nature.com/articles/s41598-021-00827-0]
- 69. Lamp Assays - TekTalk Newsletter [https://www.agilent.com/about/tektalk/en/newsletter-lamp-assays.html?srsltid=AfmBOop146iVNrnPJkZP6icApEE19sZyUzd-O-MzJaYStTxL9vx7L0o1]
- 70. Advancements in LAMP-Based Diagnostics [https://www.mdpi.com/1422-0067/25/21/11506]
- 71. RT-LAMP assay for rapid detection of the R203M mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8982466/]
- 72. Development and clinical validation of loop-mediated ... [https://www.sciencedirect.com/science/article/pii/S1198743X2100149X]
- 73. LAMP Technology for Viral Pathogen Research and ... [https://www.thermofisher.com/us/en/home/products-and-services/promotions/life-science/lamp-technology-viral-pathogen-research-surveillance.html]
- 74. Development of PCR, qPCR and LAMP methods for the ... [https://www.sciencedirect.com/science/article/pii/S2405676625000381]
- 75. Accessibility - Color usage and contrast [https://service.tamu.edu/TDClient/36/Portal/KB/PrintArticle?ID=848]
- 76. A Manually driven Centrifugal Microfluidic-LAMP Platform for ... [https://sciety-labs.elifesciences.org/articles/by?article_doi=10.21203/rs.3.rs-7032792/v1]
- 77. Getting started with loop-mediated isothermal amplification ... [https://www.neb.com/en-us/nebinspired-blog/getting-started-with-loop-mediated-isothermal-amplification?srsltid=AfmBOoomOwck7244RrMj6FuNlj7qb4dO487HsCjfir8a1aIjV5y-Blcl]
- 78. Diverse methods of reducing and confirming false-positive ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267023009145]
- 79. Electrostatically Adjustable AuNR-Mediated Thermal-Start ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12444552/]
- 80. A lyophilized open-source RT-LAMP assay for molecular ... [https://www.life-science-alliance.org/content/8/10/e202403167]
- 81. Reduced False Positives and Improved Reporting of Loop ... [https://www.nature.com/articles/s41598-019-43817-z]
- 82. Loop-mediated isothermal amplification allows testing to ... [https://www.neb.com/en-us/nebinspired-blog/loop-mediated-isothermal-amplification-allows-testing-to-be-performed-anywhere?srsltid=AfmBOor9vYMEkUPNuD18ZeHpPUOQRYiyVpqucZmSoyyE2xRjNyFyyzqS]
- 83. Inhibition of Non-specific Amplification in Loop-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9326409/]
- 84. Inhibitors of LAMP used to detect Tenacibaculum sp. strain ... [https://www.sciencedirect.com/science/article/pii/S0167701224000988]
- 85. Loop-Mediated Isothermal Amplification [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/isothermal-amplification/loop-mediated-isothermal-amplification-lamp?srsltid=AfmBOoqb8rdBA2gcfgq2RJvs8bN8-KtkS7Qas47A6hWD3rG3kaBFHADO]
- 86. A comprehensive guide to loop-mediated isothermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12142221/]
- 87. Optimization of Loop-Mediated Isothermal Amplification ... [https://www.mdpi.com/1422-0067/26/13/5933]
- 88. Optimization of a Loop Mediated Isothermal Amplification ... [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2019.00067/full]
- 89. Real-time Detection and Monitoring of Loop Mediated ... [https://www.nature.com/articles/s41598-018-23930-1]
- 90. Optimization of a loop-mediated isothermal amplification ... [https://www.sciencedirect.com/science/article/pii/S1056617124001119]
- 91. Optimization of Loop-Mediated Isothermal Amplification for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560903/]
- 92. The Sensitivity and Specificity of Loop-Mediated Isothermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8826321/]
- 93. Diagnostic accuracy of low-complexity, manual nucleic acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12510911/]
- 94. Validation of a Loop-Mediated Isothermal Amplification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7105778/]
- 95. A diagnostic accuracy study comparing RNA LAMP, direct ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.1063414/full]
- 96. Diagnostic performance of a colorimetric RT -LAMP for the ... [https://www.sciencedirect.com/science/article/pii/S2589537021003813]
- 97. a systematic review and meta-analysis [https://www.sciencedirect.com/science/article/pii/S0732889321000626]
- 98. Investigation and validation of labelling loop mediated ... [https://www.nature.com/articles/s41598-022-11320-7]
- 99. Comparative evaluation of 19 reverse transcription loop ... [https://www.nature.com/articles/s41598-020-80314-0]
- 100. Evaluation of RT-qPCR and Loop-Mediated Isothermal ... [https://www.mdpi.com/2073-4425/12/5/659]
- 101. Development and comparative assessment of RT-qPCR ... [https://www.sciencedirect.com/science/article/pii/S0048969724055906]
- 102. Development of an RT-LAMP Assay for Detecting tet(M) in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109771/]
- 103. Loop-Mediated Isothermal Amplification [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/isothermal-amplification/loop-mediated-isothermal-amplification-lamp?srsltid=AfmBOoqYWNgNEPn3U75GZQnEd9nRoT5IwtxU23mjv-ROiK82wcZ-MhlS]
- 104. Real-time optical analysis of a colorimetric LAMP assay for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8730521/]
- 105. Recent Progress on Rapid Diagnostic Methods for Viral ... [https://www.sciencedirect.com/science/article/pii/S2950248925000811]
- 106. Performance of rapid diagnostic tests, microscopy, loop ... [https://pubmed.ncbi.nlm.nih.gov/34579729/]
- 107. Analytical Method Validation: Back to Basics, Part II [https://www.chromatographyonline.com/view/analytical-method-validation-back-basics-part-ii]
- 108. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 109. Method Validation Essentials, Limit of Blank ... [https://www.biopharminternational.com/view/method-validation-essentials-limit-blank-limit-detection-and-limit-quantitation]
- 110. Virus detection by short read high throughput sequencing ... [https://www.nature.com/articles/s41541-025-01104-1]
- 111. Variation in LOD Across SARS-CoV-2 Assay Systems [https://pmc.ncbi.nlm.nih.gov/articles/PMC7798922/]
- 112. 2025 MIPS Measure #338: HIV Viral Suppression [https://mdinteractive.com/mips_quality_measure/2025-mips-quality-measure-338]
- 113. Comparative Evaluation of the LAMP Assay and PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6129767/]
- 114. a technology transfer assessment study (LAMP-TTA) during a ... [https://malariajournal.biomedcentral.com/articles/10.1186/s12936-025-05631-z]
- 115. Multisite Clinical Validation of Isothermal Amplification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8528118/]
- 116. Multi-Laboratory Validation of a Loop-Mediated Isothermal ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.00562/full]
- 117. Validation study of a novel, rapid, open platform real-time ... [https://www.nature.com/articles/s41598-025-93565-6]
- 118. Validation of quantitative loop-mediated isothermal ... [https://www.nature.com/articles/s41598-023-46001-6]
- 119. One-hour extraction-free loop-mediated isothermal ... [https://www.nature.com/articles/s41467-025-62454-x]
- 120. Method development and clinical validation of LAMP ... [https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2025.1533100/full]
- 121. Development and comparison of loop-mediated isothermal ... [https://www.sciencedirect.com/science/article/pii/S0022030219300116]
- 122. Molecular Diagnostic Tests for SARS-CoV-2 [https://www.fda.gov/medical-devices/covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2]
- 123. Reverse-Transcription Loop-Mediated Isothermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8806713/]
- 124. Evaluation of a novel direct RT-LAMP assay for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8942569/]
- 125. Evaluation of RT-LAMP Assay for Rapid Detection of SARS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9384514/]
- 126. Cross comparison of alternative diagnostic protocols ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2024.1445142/full]
- 127. A molecular test based on RT-LAMP for rapid, sensitive ... [https://www.nature.com/articles/s41598-021-95799-6]
- 128. Modeling the effectiveness of RT-PCR, RT-LAMP, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535133/]
- 129. Loop-mediated Isothermal Amplification Market 2025-2035 [https://www.futuremarketinsights.com/reports/loop-mediated-isothermal-amplification-market]
- 130. Economic Evaluation of Molecular Testing for Pulmonary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497363/]
- 131. LAMP using distance based paper device for quantitative ... [https://www.nature.com/articles/s41598-025-21371-1]
- 132. Rapid and Cost-Effective ABO Blood Genotyping Using a ... [https://www.mdpi.com/2075-4418/15/20/2568]