Nested PCR for SARS-CoV-2: A High-Sensitivity Detection Strategy for Research and Clinical Diagnostics
This article provides a comprehensive analysis of nested PCR assays for the detection of SARS-CoV-2, addressing the critical need for highly sensitive diagnostic tools in biomedical research and therapeutic development.
Nested PCR for SARS-CoV-2: A High-Sensitivity Detection Strategy for Research and Clinical Diagnostics
Abstract
This article provides a comprehensive analysis of nested PCR assays for the detection of SARS-CoV-2, addressing the critical need for highly sensitive diagnostic tools in biomedical research and therapeutic development. It explores the foundational principles that give nested PCR its enhanced sensitivity and specificity over conventional methods like qRT-PCR. The scope covers detailed methodological protocols, including primer design for stable genomic regions and one-step nested RT-PCR (OSN-qRT-PCR) workflows. It further delves into troubleshooting common challenges and optimizing assays for complex samples, such as wastewater. Finally, the article presents rigorous validation data and comparative performance analyses against other gold-standard techniques like digital droplet PCR (ddPCR) and various commercial RT-PCR kits, highlighting its superior detection rates for low viral loads and emerging variants.
The Science Behind Nested PCR: Enhancing Sensitivity for SARS-CoV-2 Detection
Within the ongoing research on SARS-CoV-2 detection, the development of highly sensitive and specific diagnostic assays remains a critical focus. Two-stage amplification techniques, such as nested and semi-nested PCR, have emerged as powerful tools to achieve this goal, particularly when detecting low viral loads or in complex sample matrices. These methods significantly enhance assay performance by adding a second, internal amplification step, which increases both the quantity and fidelity of the target amplicon. This application note details the core principles of two-stage amplification, provides a quantitative comparison of its performance, and outlines detailed protocols for its implementation in SARS-CoV-2 research, providing a valuable resource for scientists and drug development professionals.
Core Principles and Performance Advantages
Two-stage amplification methods function on a simple yet powerful principle: the product of an initial amplification reaction serves as the template for a second, internal reaction. This design confers significant advantages in sensitivity and specificity.
Enhanced Sensitivity: The second amplification stage selectively enriches the target sequence, enabling the detection of very low copy numbers that might fall below the detection limit of single-step assays. The initial amplification round increases the template concentration, while the second round, using primers that bind within the first amplicon, ensures exponential amplification of the specific target. Research has demonstrated that nested PCR can detect SARS-CoV-2 RNA at concentrations as low as 0.015 ng/μL [1]. Another study reported a limit of detection (LOD) of 7.2 copies/reaction for a semi-nested RT-PCR assay, significantly boosting the ability to identify infections with low viral loads [2]. Similar approaches using nested RT-LAMP have achieved an LOD of 5 copies/μL, outperforming standard RT-LAMP and RT-PCR [3].
Superior Specificity: The requirement for two distinct primer sets to bind correctly to the target sequence in successive reactions drastically reduces the potential for non-specific amplification and false-positive results. If the first round amplifies a non-specific product, it is highly unlikely that the internal primers will find a binding site within it. This dual verification process results in exceptionally high specificity. Multiple studies on SARS-CoV-2 nested PCR have reported specificities of 100% [1] [4]. This high specificity is also maintained in novel isothermal methods like nested RPA, which showed no cross-reactivity with other common respiratory pathogens [5].
Robustness against Sequence Variation: Targeting multiple conserved regions within the viral genome makes these assays more resilient to mismatches caused by viral mutation. A heptaplex (7-plex) semi-nested RT-PCR was designed to target seven conserved genomic regions, making it more reliable in the face of evolving viral variants [2].
Adaptability to High-Throughput and Point-of-Care Testing: The high sensitivity of two-stage amplification enables efficient sample pooling strategies without significant loss of sensitivity, facilitating large-scale screening [2]. Furthermore, innovations like fully integrated cartridges and one-tube nested RPA 2.0 are making these sensitive assays viable for rapid, equipment-light point-of-care testing [5] [3].
Table 1: Performance Comparison of SARS-CoV-2 Detection Methods
| Methodology | Reported Sensitivity | Reported Specificity | Limit of Detection (LOD) | Key Advantage |
|---|---|---|---|---|
| Conventional Nested PCR [1] [4] | 95% - 100% | 100% | 0.015 ng/μL; ~50 copies/μL | Cost-effective; high sensitivity for low viral loads |
| Semi-Nested RT-PCR [2] | 100% | 99.87% | 7.2 copies/reaction | Enables high-throughput pooled testing |
| Two-Step SYBR Green RT-qPCR [6] | 88% (N & S genes combined) | 86% (N & S genes combined) | Up to 1:106 dilution | Lower cost than one-step probe-based methods |
| One-Step RT-qPCR (TaqMan) [7] [8] | Reference Standard | Reference Standard | Varies by kit | High throughput; widely adopted gold standard |
| Nested RT-LAMP [3] | 100% | 98% | 5 copies/μL | Isothermal; visual detection; high sensitivity |
| Nested RPA [5] | Comparable to RT-qPCR | High (no cross-reactivity) | 2 copies/reaction | Rapid (≤30 min); low-temperature isothermal |
The following workflow illustrates the general procedure for a two-stage amplification assay, from sample preparation to final detection:
Detailed Experimental Protocol: Nested PCR for SARS-CoV-2
This protocol is adapted from a study that developed and validated a conventional nested PCR targeting the SARS-CoV-2 N gene, demonstrating 100% sensitivity and specificity [1].
Reagents and Equipment
Table 2: Research Reagent Solutions
| Item | Function / Application | Example / Specification |
|---|---|---|
| ISOLATE II RNA Mini Kit | Viral RNA extraction from swab samples | (Bioline) [1] |
| SensiFAST cDNA Synthesis Kit | Reverse transcription of RNA to cDNA | (Bioline) [1] |
| My Taq HS Red Mix | DNA polymerase for PCR amplification | (Bioline) [1] |
| External Primers | First-round amplification of the target gene | Ext2019nCorVF/VR [1] |
| Internal Primers | Second-round amplification for specificity | intF/intR [1] |
| Thermal Cycler | Platform for PCR amplification | e.g., QB96, SaCycler-96 [1] |
| Agarose Gel Electrophoresis System | Analysis of PCR amplicon size and quality | 2% agarose gel [1] |
Primer Sequences
The following primers were designed based on the N gene of SARS-CoV-2 (GenBank sequence MN908947.3) [1]:
- External Forward (Ext2019nCorVF): 5′-GGCAGTAACCAGAATGGAGA-3′
- External Reverse (Ext2019nCorVR): 5′-CTCAGTTGCAACCCATATGAT-3′
- Product Size: 335 bp
- Internal Forward (intF): 5′-CACCGCTCTCACTCAACAT-3′
- Internal Reverse (intR): 5′-CATAGGGAAGTCCAGCTTCT-3′
- Product Size: 212 bp
Step-by-Step Procedure
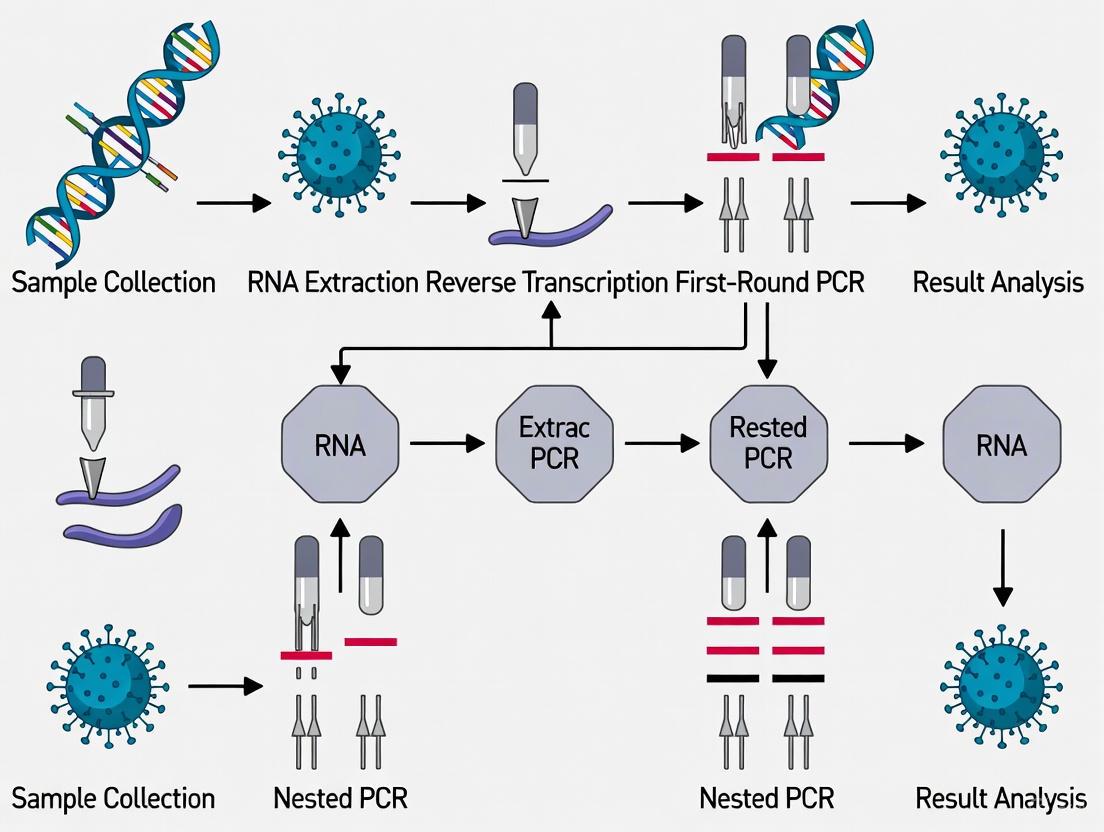
RNA Extraction and Reverse Transcription:
- Extract viral RNA from nasopharyngeal, oropharyngeal, or other swab samples using a commercial RNA extraction kit, following the manufacturer's instructions.
- Perform reverse transcription (RT) to synthesize cDNA. A typical 20 μL reaction contains: 7 μL of extracted RNA, 1 μL of RT enzyme, 4 μL of TransAmp buffer, and 8 μL of DEPC-treated water.
- Use the following thermocycler conditions for RT: 25°C for 10 min, 42°C for 15 min, and 80°C for 5 min.
First Round of Nested PCR:
- Prepare the first PCR reaction mix on ice. A 25 μL reaction contains:
- 12.5 μL of My Taq HS Red Mix
- 1 μL of external forward primer (10 pmol/μL)
- 1 μL of external reverse primer (10 pmol/μL)
- 4 μL of cDNA template
- 6.5 μL of PCR-grade water
- Run the first PCR amplification using the following cycling parameters:
- Initial Denaturation: 95°C for 1 min
- Amplification (35 cycles): 95°C for 15 s, 54.6°C for 15 s, 72°C for 10 s
- Final Extension: 72°C for 1 min
- Prepare the first PCR reaction mix on ice. A 25 μL reaction contains:
Second Round of Nested PCR:
- Prepare the second PCR reaction mix on ice. A 25 μL reaction contains:
- 12.5 μL of My Taq HS Red Mix
- 1 μL of internal forward primer (10 pmol/μL)
- 1 μL of internal reverse primer (10 pmol/μL)
- 0.5 μL of the first-round PCR product (dilution may be optimized)
- 10 μL of PCR-grade water
- Run the second PCR amplification using the same cycling parameters as the first round.
- Prepare the second PCR reaction mix on ice. A 25 μL reaction contains:
Detection and Analysis:
- Analyze 5-10 μL of the second-round PCR product by 2% agarose gel electrophoresis.
- Visualize the gel using a UV transilluminator. A positive result is confirmed by the presence of a band at the expected size of 212 bp.
- For absolute confirmation, the amplified product can be purified and subjected to Sanger sequencing using the internal primers [1].
Advanced Application: Semi-Nested Multiplex RT-PCR with Melting Analysis
For higher throughput and variant resilience, a semi-nested, heptaplex (7-plex) RT-PCR can be employed. This advanced protocol uses a pre-amplification step followed by a highly multiplexed real-time PCR with melting curve analysis [2].
The diagram below outlines the two major steps of this semi-nested assay, which combines pre-amplification with multiplex detection and analysis.
Key Protocol Steps
Pre-Amplification:
- Perform reverse transcription and a limited-cycle (e.g., 15-20 cycles) PCR using a primer mix targeting the seven conserved regions of the SARS-CoV-2 genome (E, N, and ORF1ab genes). This step enriches the target sequences.
Semi-Nested Multiplex RT-PCR and Melting Analysis:
- Use the pre-amplified product as the template for a multiplex real-time PCR reaction containing seven pairs of primers targeting the same regions.
- Run the reaction on a real-time PCR instrument and perform a high-resolution melting analysis after the amplification cycles.
Data Interpretation:
- The complex melting spectrum generated from the seven amplicons is analyzed using a cloud-based artificial intelligence (AI) algorithm (e.g., a gradient-boosted trees classifier) to differentiate between positive and negative samples with high accuracy [2].
Two-stage amplification is a foundational principle that reliably enhances the sensitivity and specificity of molecular diagnostics for SARS-CoV-2. The protocols detailed herein, from conventional gel-based nested PCR to advanced AI-driven multiplex assays, provide researchers with robust tools for detecting low viral loads, screening animal reservoirs, and conducting large-scale epidemiological studies. The continued innovation in this space, particularly the integration of isothermal amplification and point-of-care form factors, promises to make this powerful diagnostic principle even more accessible and impactful in the global effort to manage COVID-19 and future infectious disease threats.
The detection of pathogens with low viral loads presents a significant challenge in molecular diagnostics, particularly during the early or late stages of infection. While conventional polymerase chain reaction (PCR) is a foundational technique, its sensitivity limitations can lead to false-negative results when pathogen concentrations are minimal [9]. Nested PCR has emerged as a powerful alternative that effectively overcomes these limitations through a two-stage amplification process that dramatically enhances detection capabilities.
This technical note details the superior performance of nested PCR for detecting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), especially in samples with low viral loads, such as those from asymptomatic individuals, patients in the convalescent phase, or environmental samples like wastewater. We provide comparative quantitative data, detailed experimental protocols, and practical implementation guidelines to facilitate the adoption of this sensitive and cost-effective methodology in research and diagnostic settings.
Comparative Performance Data
Quantitative Comparison of PCR Methods
Extensive validation studies demonstrate that nested PCR consistently outperforms conventional single-round PCR and shows comparable or superior sensitivity to real-time quantitative PCR (qPCR), particularly at low target concentrations.
Table 1: Comparative Sensitivity of PCR Methods for Pathogen Detection
| Pathogen | Detection Method | Sensitivity | Limit of Detection | Key Advantage |
|---|---|---|---|---|
| SARS-CoV-2 [10] [4] | Nested PCR (N gene) | 95% | ~50 copies/µL (Ct 31.5) | Cost-effective for large-scale animal surveillance |
| SARS-CoV-2 [11] | One-Step Nested qRT-PCR (ORF1ab) | 82.35% (vs. 58.82% for qRT-PCR) | 194.74 copies/mL | Superior clinical detection rate in patient samples |
| SFTS Virus [12] | RT-Nested PCR (M segment) | 100% (initial samples) | N/A | Detection up to 40 days post-symptom onset |
| SARS-CoV-2 [13] | Nested PCR + NGS | N/A | Effective in inhibitor-rich wastewater | Enables variant tracking in low-concentration environmental samples |
Direct Sensitivity Comparison
A prospective clinical study on Severe Fever with Thrombocytopenia Syndrome (SFTS) virus provides a direct, head-to-head comparison of different PCR methods, illustrating a common pattern of performance that applies to SARS-CoV-2 detection as well.
Table 2: SFTSV Detection Rate in Patient Samples Over Time [12]
| Days Post-Symptom Onset | Single-Round PCR-M | Real-Time QPCR-S | Nested PCR-S | Nested PCR-M |
|---|---|---|---|---|
| 1-7 days | 63% | 92% | 92% | 97% |
| 8-21 days | 44% | 71% | 75% | 85% |
| 22-40 days | Marked decrease | Marked decrease | ~70% | ~70% |
| Overall Positivity | 44% | 71% | 75% | 85% |
This data demonstrates that nested PCR maintains a high detection rate even in the convalescent phase when viral loads diminish, a critical period where conventional methods often fail.
Principles and Mechanisms
Technical Workflow
The fundamental advantage of nested PCR lies in its two-stage amplification process. The first PCR round uses an outer primer pair to amplify the target sequence. A small aliquot of this product is then transferred to a second reaction containing an inner primer pair that binds within the first amplicon, resulting in exponential sensitivity enhancement.
Key Advantages for Low Viral Load Detection
This two-stage process confers several critical advantages for challenging samples:
- Enhanced Sensitivity: The second round of amplification effectively increases the total cycle number without excessive background, enabling detection of very low copy numbers (as low as 50 copies/µL for SARS-CoV-2) [10] [4].
- Improved Specificity: The requirement for two independent primer pairs to bind correctly significantly reduces false positives from non-specific amplification or primer-dimer artifacts [11].
- Tolerance to Inhibitors: Samples often contain substances that inhibit PCR. The dilution step between rounds reduces the concentration of these inhibitors in the second reaction, improving robustness [13].
Application Notes: SARS-CoV-2 Detection
Detailed Protocol for SARS-CoV-2 N Gene Detection
The following protocol, adapted from Panei et al. (2024), provides a optimized workflow for detecting SARS-CoV-2 in animal and human samples [10] [4].
Sample Preparation and RNA Extraction
- Sample Type: Oropharyngeal swabs suspended in viral transport media.
- RNA Extraction: Use membrane adsorption kits (e.g., from Di'an, Hangzhou, China) following manufacturer's instructions [11].
- Reverse Transcription: Convert 10 µL of extracted RNA to cDNA using a High-Capacity cDNA Reverse Transcription Kit with random hexamers.
First Round PCR Amplification
- Reaction Mix:
- 5 µL cDNA template
- 5 pmol/µL each outer forward and reverse primer
- 12.5 µL 2X PCR Master Mix
- Nuclease-free water to 25 µL
- Outer Primers (targeting SARS-CoV-2 N gene):
- Forward:
5'-GCCGCATTACGTTTGGTGGAC-3' - Reverse:
5'-GCGAGGTCTGTTACAAGCTTG-3'(produces 633-bp fragment)
- Forward:
- Cycling Conditions:
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: 49°C for 40 seconds
- Extension: 72°C for 45 seconds
- Final extension: 72°C for 5 minutes
Second Round PCR Amplification
- Reaction Mix:
- 2 µL of 1:50 dilution of first-round product
- 5 pmol/µL each inner forward and reverse primer
- 12.5 µL 2X PCR Master Mix
- Nuclease-free water to 25 µL
- Inner Primers (nested within first amplicon):
- Forward:
5'-CGAATGGCTGTTTACCGCGCA-3' - Reverse:
5'-GGTCCGCCACATAATCGATCC-3'(produces 248-bp fragment)
- Forward:
- Cycling Conditions:
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: 51°C for 40 seconds
- Extension: 72°C for 30 seconds
- Final extension: 72°C for 5 minutes
Detection and Analysis
- Analyze 10 µL of the second-round product by agarose gel electrophoresis (2% gel).
- Visualize bands under UV transillumination after ethidium bromide staining.
- The expected 248-bp band indicates a positive SARS-CoV-2 detection.
Research Reagent Solutions
Table 3: Essential Reagents for Nested PCR Detection of SARS-CoV-2
| Reagent/Category | Specific Examples | Function & Application Note |
|---|---|---|
| Nucleic Acid Extraction | Membrane adsorption kits (e.g., Di'an, Hangzhou) [11] | Purifies RNA from swab samples; critical for removing PCR inhibitors |
| Reverse Transcription | High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) [14] | Converts viral RNA to stable cDNA for amplification |
| PCR Amplification | Platinum SuperFi II PCR Master Mix (Thermo Fisher) [14] | High-fidelity enzyme mix for specific target amplification |
| Primers (Outer) | Custom synthesized, targeting N gene [10] | First amplification pair; should flank a 500-700 bp region |
| Primers (Inner) | Custom synthesized, nested within outer product [10] | Second amplification pair; critical for specificity and sensitivity |
| Electrophoresis | Agarose, ethidium bromide, DNA ladder | Confirms amplicon size and reaction specificity |
Adaptation for Wastewater Surveillance
For environmental samples like wastewater with extremely low viral concentrations and high inhibitor content, the protocol requires modification [13]:
- Sample Concentration: Centrifuge wastewater samples at 15,000 × g for 1.5 hours before RNA extraction.
- Inhibition Management: Include a 1:10 dilution step of the first-round product to reduce inhibitor carryover.
- Target Selection: Amplify smaller fragments (∼230 bp) of the Spike gene to accommodate potentially degraded RNA.
- Downstream Application: Purify nested PCR products for next-generation sequencing to track variant circulation.
Technical Considerations
Contamination Prevention
The high sensitivity of nested PCR increases vulnerability to amplicon contamination. Implement these stringent controls:
- Physical Separation: Perform pre-PCR (setup), first-round, and second-round PCR in separate dedicated areas.
- Dedicated Equipment: Use separate pipettes, tips, and reagents for each stage.
- Negative Controls: Include multiple negative controls (no-template and no-reverse transcription) across both amplification rounds.
- UV Irradiation: Expose workstations to UV light between procedures to degrade contaminating DNA.
Validation and Quality Control
- Analytical Sensitivity: Determine the limit of detection (LoD) using serial dilutions of quantified RNA standards [10].
- Specificity Testing: Validate primer specificity against related pathogens (e.g., canine coronavirus, feline infectious peritonitis virus for SARS-CoV-2) [10] [4].
- Inhibition Assessment: Spike samples with known quantities of synthetic control RNA to detect inhibition.
- Concordance Testing: Compare results with established qPCR methods to calculate kappa coefficient of agreement [10].
Nested PCR represents a highly effective solution for overcoming the critical challenge of low viral load detection in SARS-CoV-2 research and surveillance. Its enhanced sensitivity and specificity, combined with lower operational costs compared to real-time PCR, make it particularly valuable for large-scale animal studies [10] [4], wastewater monitoring [13], and clinical detection in the convalescent phase [12].
While requiring careful contamination control, the technique provides researchers with a powerful, accessible tool for tracking virus circulation and understanding infection dynamics, especially in resource-limited settings. The protocols and data presented herein provide a foundation for implementing this robust methodology in diverse research applications.
The rapid evolution of SARS-CoV-2 and the emergence of variants with signature mutations pose a significant challenge to the diagnostic accuracy of molecular assays, particularly those based on specific genomic targets. Mutations in primer and probe binding regions can lead to mismatches, resulting in diminished amplification and potentially false-negative results [15]. This application note details a structured in silico methodology for the evaluation of three key SARS-CoV-2 genes—N, ORF1ab, and ORF3a—to identify the most stable and suitable target for a robust nested PCR assay. This work is situated within a broader thesis research project aimed at developing highly reliable and cost-effective diagnostic tools for SARS-CoV-2 detection, especially in resource-limited settings. The protocols described herein provide a framework for researchers and drug development professionals to systematically select genomic targets that are less prone to escape detection due to viral evolution.
In Silico Analysis Protocol for Target Gene Evaluation
- Sequence Databases: Access the GISAID EpiCoV database and the NCBI Virus database for the most current and comprehensive collection of SARS-CoV-2 genomic sequences.
- Bioinformatics Tools: Use tools like NCBI Primer-BLAST for initial specificity checks and Oligo 7 software for advanced primer design and analysis.
- Alignment and Analysis Software: MUSCLE or MAFFT for multiple sequence alignments; custom Python or R scripts for mutation frequency analysis.
Step-by-Step Workflow
- Sequence Retrieval: Download a curated dataset of complete, high-coverage SARS-CoV-2 genomes, ensuring representation of all major Variants of Concern (VOCs) over time.
- Target Region Extraction: Isolate the genomic sequences corresponding to the N, ORF1ab, and ORF3a genes from the whole-genome records.
- Multiple Sequence Alignment: Perform a multiple sequence alignment for each gene set to identify conserved regions and single nucleotide polymorphisms (SNPs).
- Mutation Frequency Analysis: Calculate the mutation frequency for each gene by analyzing the aligned sequences for variations relative to the reference genome (e.g., MN908947). The frequency can be expressed as the number of unique sequences containing a mutation divided by the total number of sequences analyzed.
- Variant Cross-Reference: Cross-reference identified mutations with known VOC-defining mutations to assess their prevalence and potential impact on public health surveillance.
- Conserved Region Identification: Pinpoint regions of high conservation (minimal mutations across all analyzed sequences) suitable for primer design.
Diagram: The following workflow illustrates the sequential protocol for the in silico analysis:
Comparative Genomic Stability and Primer Design
Quantitative Stability Assessment
A comparative analysis of the N, ORF1ab, and ORF3a genes reveals significant differences in their stability and mutation profiles, which directly impacts their suitability as diagnostic targets.
Table 1: Comparative Gene Stability and Mutation Profile
| Gene | Primary Function | Reported Mutations | Key Stability Findings |
|---|---|---|---|
| N Gene | Structural protein; forms viral capsid [16] | Lower mutation frequency; more conserved [4] | Highly conserved and stable; fewer mutations described [4] [17]. Higher amino acid homology across variants [4]. Recommended as a primary target. |
| ORF1ab | Encodes non-structural proteins for replication | Multiple unique mutations reported (e.g., C11450A, C14178T) [15] | Prone to mutations causing diagnostic escape [15]. A variant with 5 unique mutations led to a ~10 Ct value discrepancy in a commercial dual-target assay [15]. Use as a secondary target only with careful surveillance. |
| ORF3a | Accessory protein; involved in virulence and egress [18] | Functional studies focus on protein function, not diagnostic stability [18] | Limited direct evidence on sequence stability for diagnostics. Its role in driving dynamic dense body formation for optimal viral infectivity is established [18]. Requires further stability validation for diagnostic use. |
Validated Primer Sequences for Nested PCR
Based on the stability analysis, the N gene is the most suitable target. The following primer sets have been empirically validated for sensitivity and specificity in nested PCR assays.
Table 2: Validated Primer Sequences for N Gene Nested PCR [4] [17]
| Primer Name | Sequence (5' → 3') | Amplification Round | Product Length |
|---|---|---|---|
| NFExternal | ACAACAGAACGGAAAGCAAC | First | 633 bp [4] |
| NRExternal | GGACAGCATCAGTAGCAATC | First | 633 bp [4] |
| NFInternal | GGAACCACTAGTGCCAGTTG | Second | 358 bp [4] |
| NRInternal | CCAACACCAGCACCATTATC | Second | 358 bp [4] |
Experimental Validation Protocol for Nested PCR
Sample Preparation and RNA Extraction
- Sample Collection: Collect nasopharyngeal/oropharyngeal swabs and place them in Viral Transport Media (VTM).
- RNA Extraction: Use a magnetic bead-based nucleic acid extraction kit, such as the ISOLATE II RNA Mini Kit. Elute the final RNA pellet in 50 μL of RNase-free water [17].
Reverse Transcription and Nested PCR Amplification
This protocol is adapted from established methods with demonstrated high sensitivity and specificity [4] [17].
Reverse Transcription (RT):
- Reaction Setup: Combine 7 μL of extracted RNA, 8 μL of DEPC-treated water, 4 μL of TransAmp buffer, and 1 μL of reverse transcriptase enzyme.
- Thermocycling Conditions: Incubate at 25°C for 10 minutes, followed by 42°C for 15 minutes, and a final inactivation step at 80°C for 5 minutes. The resulting cDNA can be stored at -10°C [17].
First Round of PCR:
- Reaction Mix: 12.5 μL of 2x My Taq HS Red Mix, 1 μL of each external primer (10 pmol/μL), 4 μL of cDNA template, and 6.5 μL of PCR-grade water to a final volume of 25 μL.
- Thermocycling Conditions: Initial denaturation at 95°C for 1 min; followed by 35 cycles of denaturation at 95°C for 15 sec, annealing at 49°C for 15 sec, and extension at 72°C for 15 sec; with a final extension at 72°C for 1 min [4].
Second Round of PCR (Nested):
- Reaction Mix: 12.5 μL of 2x My Taq HS Red Mix, 1 μL of each internal primer (10 pmol/μL), 0.5 μL of the first-round PCR product, and 10 μL of PCR-grade water to a final volume of 25 μL.
- Thermocycling Conditions: Use the same cycling conditions as the first round, but with an annealing temperature of 51°C [4].
Product Analysis and Assay Validation
- Gel Electrophoresis: Analyze 5-10 μL of the second-round PCR product on a 2% agarose gel stained with ethidium bromide. A positive result is indicated by a clear band at the expected size (358 bp) [17].
- Sequencing (Optional): Purify the PCR product and perform Sanger sequencing using the internal primers to confirm the target sequence [17].
Diagram: The nested PCR process involves two consecutive amplification rounds to enhance specificity and sensitivity:
Performance Metrics and The Scientist's Toolkit
Assay Performance Characteristics
When validated against reference real-time RT-PCR methods, the nested PCR assay targeting the N gene demonstrates excellent performance, particularly in detecting low viral loads.
Table 3: Performance Metrics of the N Gene Nested PCR Assay
| Performance Parameter | Result | Experimental Note |
|---|---|---|
| Analytical Sensitivity (LoD) | ~50 copies/μL [4] | Corresponds to a Ct value of approximately 31.5 from a real-time RT-PCR assay [4]. |
| Clinical Sensitivity | 95-100% [4] [17] | Achieved 100% detection of positive samples in a clinical validation [17]. |
| Clinical Specificity | 100% [4] [17] | No cross-reactivity with other common coronaviruses (e.g., CCoV, FIPV) or respiratory pathogens [4] [17]. |
| Agreement (Kappa Value) | 0.829 (Excellent) [4] | Indicates a very high level of agreement with the reference real-time RT-PCR method [4]. |
Research Reagent Solutions
Table 4: Essential Materials and Reagents for Nested PCR Assay
| Item | Function / Application | Example Product / Note |
|---|---|---|
| RNA Extraction Kit | Purification of viral RNA from clinical samples (swabs, sputum). | Magnetic bead-based kits (e.g., ISOLATE II RNA Mini Kit, apsLABS Viral Nucleic Acid Extraction Kit) [19] [17]. |
| cDNA Synthesis Kit | Reverse transcription of purified RNA into stable cDNA for PCR amplification. | Kits containing reverse transcriptase and buffer (e.g., SensiFAST cDNA Synthesis Kit) [17]. |
| PCR Master Mix | Provides optimal buffer, dNTPs, and a thermostable DNA polymerase for robust amplification. | HS PCR mixes (e.g., My Taq HS Red Mix) [17]. |
| Validated Primers | Specifically target and amplify conserved regions of the SARS-CoV-2 N gene. | See Table 2 for validated sequences [4] [17]. |
| Positive Control | Validates the entire workflow, from extraction to amplification. | Inactivated SARS-CoV-2 isolate (e.g., USA-WA1/2020) [17]. |
| Agarose Gel System | Visualization and confirmation of the amplified PCR product. | Standard equipment for gel electrophoresis [17]. |
The adaptive immune response to Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) involves the coordinated activation of both cellular and humoral immunity. The humoral response, characterized by the production of immunoglobulins (Ig) against viral antigens, provides crucial protection and serves as a marker of prior exposure. This application note details methodologies for investigating the relationship between active SARS-CoV-2 infection, as determined by PCR positivity, and the subsequent seroprevalence of virus-specific IgG antibodies. Framed within broader research on nested PCR assays for SARS-CoV-2 detection, this protocol provides researchers and drug development professionals with standardized procedures for correlating molecular diagnostic data with serological outcomes, which is essential for understanding infection dynamics, immune persistence, and vaccine efficacy.
The relationship between PCR-confirmed infection and subsequent IgG seroprevalence has been quantified in multiple recent studies. The data below summarize key findings on antibody persistence and detection rates.
Table 1: IgG Seroprevalence Following PCR-Confirmed SARS-CoV-2 Infection
| Study Cohort / Context | Time Post-PCR Positive | Anti-Spike IgG Seroprevalence | Anti-Nucleocapsid IgG Seroprevalence | Key Correlates |
|---|---|---|---|---|
| Norwegian Cohort (n=400 PCR+) [20] | 12 months | 97% (278/287) | 86% (248/287) | Hybrid immunity common |
| 24 months | 100% (233/233) | 95% (221/233) | Booster vaccination increased antibody levels | |
| Nigerian Hospital Cohort (n=250) [21] | Not Specified (Cross-sectional) | 41.6% (Overall, by rapid test) | Not Reported | Significantly higher in patients vs. healthcare workers |
| Polish Hospital Population (n=3,104) [22] | 2021-2023 (Retrospective) | Significantly higher in individuals >60 years old | Not Reported | Number of infections correlated with number of tests |
Table 2: Impact of Vaccination on Humoral Immune Parameters
| Parameter | Finding | Study |
|---|---|---|
| Effect of Booster Vaccination | Booster-immunized participants were 3.7x more likely to have high anti-spike antibody levels. | Norwegian Cohort [20] |
| Response in Vulnerable Populations | People Who Use Drugs (PWUD) had binding and neutralizing antibody levels comparable to controls after second vaccine dose. | Oslo, Norway Study [23] |
| Vaccination Coverage | 84% of PWUD received at least one dose, compared to 89% in the general population. | Oslo, Norway Study [23] |
Experimental Protocols
Protocol 1: Detection of Active Infection via Nested PCR
This protocol, adapted for SARS-CoV-2 detection from clinical samples, offers high sensitivity and is particularly useful for detecting low viral loads or in resource-limited settings [24] [4] [17].
1. Sample Collection and RNA Extraction
- Sample Type: Collect nasopharyngeal/oropharyngeal swabs and place in viral transport medium.
- RNA Extraction: Use a commercial RNA extraction kit (e.g., ISOLATE II RNA Mini kit). Elute RNA in 50 μL RNase-free water.
- Quality Control: Quantify and assess RNA purity using a spectrophotometer (e.g., NanoDrop 2000). Store extracts at -80°C if not used immediately.
2. Reverse Transcription (RT)
- Reaction Setup: In a nuclease-free tube, combine:
- 7 μL of extracted RNA
- 8 μL of DEPC-treated water
- 4 μL of TransAmp buffer (or equivalent from a commercial kit)
- 1 μL of reverse transcriptase enzyme
- Thermocycling Conditions:
- 25°C for 10 minutes (annealing)
- 42°C for 15 minutes (elongation)
- 80°C for 5 minutes (enzyme inactivation)
- Product Storage: Store synthesized cDNA at -20°C or proceed directly to PCR.
3. Nested PCR Amplification
- Primer Design: Design external and internal primer pairs targeting a conserved region of the SARS-CoV-2 genome, such as the N gene [4] [17].
- First Round PCR:
- Reaction Mix:
- 12.5 μL of 2x My Taq HS Red Mix
- 1 μL of each external primer (10 pmol/μL)
- 4 μL of cDNA template
- 6.5 μL of PCR-grade water
- Thermocycling Conditions:
- Initial denaturation: 95°C for 1 min
- 35 cycles of: 95°C for 15 sec, 49°C for 15 sec, 72°C for 15 sec
- Final extension: 72°C for 1 min
- Reaction Mix:
- Second Round (Nested) PCR:
- Reaction Mix:
- 12.5 μL of 2x My Taq HS Red Mix
- 1 μL of each internal primer (10 pmol/μL)
- 0.5 μL of product from the first PCR
- 10 μL of PCR-grade water
- Thermocycling Conditions:
- Initial denaturation: 95°C for 1 min
- 35 cycles of: 95°C for 15 sec, 51°C for 15 sec, 72°C for 15 sec
- Final extension: 72°C for 1 min
- Reaction Mix:
4. Analysis and Validation
- Gel Electrophoresis: Separate 5 μL of the final PCR product on a 2% agarose gel stained with ethidium bromide. Visualize under UV light.
- Expected Result: A clear band of the expected size (e.g., ~633 bp for the N gene amplicon described in [4]).
- Sequencing (Optional): Purify PCR products and perform Sanger sequencing with the internal primers for definitive confirmation [17].
- Assay Validation: This method has demonstrated a sensitivity of 95% and specificity of 100% when validated against real-time RT-PCR, with a limit of detection near 50 copies/μL [4].
Protocol 2: Quantifying IgG Seroprevalence via Immunoassay
This protocol outlines the procedure for measuring SARS-CoV-2 specific IgG antibodies in serum, which indicates past infection or vaccination.
1. Sample Collection
- Collect venous blood into a clotting activator tube (e.g., 4.9 mL S-Monovette).
- Centrifuge at 3000 rpm for 10 minutes to separate serum.
- Aliquot and store serum at -80°C until analysis.
2. IgG Detection by Chemiluminescence Immunoassay (CLIA)
- Principle: Use an automated analyzer (e.g., Atellica IM Analyzer or COBAS e411) and commercial kits.
- Target Antigens: Assays typically detect IgG against the Spike (S) protein, Receptor Binding Domain (RBD), or Nucleocapsid (N) protein.
- Procedure:
- Follow manufacturer's instructions for the specific kit.
- Briefly, the assay uses a "sandwich" principle where SARS-CoV-2 antigens are bound to magnetic particles or a plate.
- Diluted patient serum is added. If present, anti-SARS-CoV-2 IgG antibodies bind to the antigens.
- After washing, a chemiluminescent-labeled anti-human IgG antibody is added.
- Trigger solutions are added, and the emitted light is measured as Relative Light Units (RLUs), which is proportional to the amount of antibody present.
- Data Interpretation: Results are reported as quantitative values (e.g., BAU/mL) after conversion using manufacturer-provided formulas. A value above the kit's specified cutoff (e.g., index value ≥1.00 or BAU/mL ≥ critical threshold) is considered positive [22].
Visualized Workflows and Relationships
Diagram 1: Experimental workflow for correlating PCR and serology data.
Diagram 2: Temporal relationship between PCR positivity and IgG response.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for SARS-CoV-2 Humoral Immune Research
| Reagent/Material | Function/Application | Example Product/Note |
|---|---|---|
| Viral Transport Medium (VTM) | Preservation of viral RNA integrity in swab samples during transport and storage. | Commercially available VTM tubes. |
| RNA Extraction Kit | Isolation of high-purity viral RNA from clinical samples for downstream molecular assays. | ISOLATE II RNA Mini Kit (Bioline) [17]. |
| Nested PCR Primers | Specific amplification of SARS-CoV-2 genomic regions in two successive rounds for enhanced sensitivity. | Custom primers targeting N gene or S gene [24] [4]. |
| Taq DNA Polymerase | Enzyme for PCR amplification of cDNA targets. | My Taq HS Red Mix (Bioline) [17]. |
| cDNA Synthesis Kit | Reverse transcription of viral RNA into stable complementary DNA (cDNA) for PCR. | SensiFAST cDNA Synthesis Kit (Bioline) [17]. |
| SARS-CoV-2 IgG CLIA Kit | Quantitative detection of human IgG antibodies against SARS-CoV-2 antigens in serum/plasma. | Assays targeting S protein RBD (e.g., Siemens Atellica IM) [22]. |
| Reference SARS-CoV-2 RNA | Positive control for validating PCR assay sensitivity and specificity. | Inactivated isolate (e.g., USA-WA1/2020) [17]. |
Implementing Nested PCR: Protocols for Human, Animal, and Environmental Surveillance
Primer Design Strategies for the N Gene and ORF1ab Region
The accurate detection of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) remains a cornerstone of effective public health response to the COVID-19 pandemic. Reverse transcription-polymerase chain reaction (RT-PCR) serves as the gold standard method, with the nucleocapsid (N) gene and ORF1ab region representing the most frequently targeted genomic areas due to their high conservation and expression levels [25] [26]. However, the rapid emergence of novel variants with mutations in primer binding sites has highlighted critical vulnerabilities in diagnostic assays, including false-negative results and reduced sensitivity [25] [26]. This application note examines advanced primer design strategies that enhance the robustness of SARS-CoV-2 detection against the evolving viral landscape, with particular emphasis on nested PCR applications that offer superior sensitivity for detecting low viral loads [27] [4] [11].
Primer Design Strategies for Conserved Target Regions
Structure-Based Primer Design for the N Gene
Conventional primer design focuses primarily on sequence conservation, but structure-based strategies offer enhanced resilience against viral evolution. This approach leverages protein structural constraints to identify genomic regions where mutations would prove functionally deleterious to the virus, thereby minimizing the risk of diagnostic escape variants.
- Core Principle: Position the 3' end of primers at codons encoding tryptophan residues within the structural core of the nucleocapsid protein. Tryptophan is encoded by a single codon (UGG), and any mutation at this position would necessarily alter the amino acid, potentially destabilizing the protein structure and reducing viral fitness [25].
- Experimental Validation: Primers designed to target tryptophan codons in the N gene demonstrated equivalent specificity and sensitivity to CDC-approved assays while theoretically eliminating the risk of variant escape through lethal structural consequences [25].
- Practical Advantage: This strategy is particularly valuable for the N gene, which has accumulated the highest number of mutations in primer and probe targets despite being one of the most conserved regions in the SARS-CoV-2 genome [25].
Deep Learning-Assisted Primer Discovery
Artificial intelligence approaches enable the identification of highly specific primer sequences without relying exclusively on sequence alignment methods, which may miss novel conserved regions.
- Methodology: Convolutional Neural Networks (CNNs) trained on coronavirus genomic sequences can identify representative 21-base pair sequences unique to SARS-CoV-2 with 98.73% classification accuracy [28].
- Validation: Sequences discovered through this method demonstrated near-perfect accuracy (>99%) in distinguishing SARS-CoV-2 from other virus strains when tested against NCBI and GISAID repositories [28].
- Implementation: The deep learning workflow analyzes filter activations from the trained network to pinpoint specific genomic sequences the model uses for classification, which can then be validated as potential primer candidates [28].
Bioinformatics Tools for Mutation Monitoring
Web-based tools specifically designed for SARS-CoV-2 primer evaluation help researchers track the impact of viral evolution on primer efficacy.
- CoVrimer Functionality: This webtool aligns user-submitted primer sequences against comprehensive SARS-CoV-2 mutation databases, visualizing mutation frequencies in primer binding sites and amplicon regions [29].
- Practical Application: Researchers can input existing or newly designed primers to identify locations with excessive mutation loads and select alternative conserved regions or incorporate degenerate bases where appropriate [29].
- Strategic Benefit: Regular monitoring of primer targets against emerging variants allows proactive assay refinement before diagnostic failures occur in clinical settings [29].
Quantitative Performance Comparison of SARS-CoV-2 Detection Methods
The following table summarizes the analytical performance of various PCR-based detection methods, highlighting the enhanced sensitivity of nested approaches:
Table 1: Performance Characteristics of SARS-CoV-2 Detection Methods
| Method | Target Genes | Limit of Detection (copies/mL) | Clinical Sensitivity | Key Applications |
|---|---|---|---|---|
| One-Step Nested qRT-PCR | ORF1ab, N | 189.1-194.7 [11] | 82.35% (28/34 samples) [11] | Low viral load detection |
| Semi-nested RT-PCR with Melting Analysis | E, N (x4), ORF1ab (x2) | 7.2 copies/reaction [2] | 100% (97.83-100% CI) [2] | High-throughput screening |
| Conventional qRT-PCR | ORF1ab, N | 520.1-528.1 [11] | 58.82% (20/34 samples) [11] | Routine diagnostic testing |
| Nested PCR (Conventional) | N | 0.015 ng/μL RNA [17] | 100% (vs. reference methods) [17] | Animal surveillance, resource-limited settings |
Table 2: Comparison of Primer Design Approaches for SARS-CoV-2 Detection
| Design Strategy | Key Principle | Advantages | Limitations |
|---|---|---|---|
| Structure-Based Design [25] | Targets structurally constrained codons | Minimizes risk of escape variants; leverages viral fitness constraints | Requires protein structure knowledge; limited to specific genomic regions |
| Deep Learning-Assisted [28] | CNN identification of unique sequences | Discovers novel conserved regions without alignment bias | Requires substantial training data; computational complexity |
| Mutation-Monitoring Tools [29] | Tracking primer mismatches in variants | Proactive assay refinement; visual mutation mapping | Reactive to existing mutations; database dependency |
Experimental Protocols
Structure-Based Primer Design and Validation Protocol
This protocol outlines the experimental workflow for designing and validating structure-based primers targeting the N gene of SARS-CoV-2.
Procedure:
Target Identification:
- Analyze the crystal structure of the SARS-CoV-2 nucleocapsid protein to identify tryptophan residues (W108, W132, W301) located in the structural core [25].
- Map these residues to their corresponding genomic coordinates in the N gene.
Primer Design:
- Design primers such that the three nucleotides at the 3' end correspond to the tryptophan codon (UGG).
- Follow standard primer design parameters (length: 18-22 bp, TM: 55-65°C, GC: 40-60%).
- Synthesize both standard primers and primers with intentional mismatches at various positions for comparison.
Specificity Validation:
- Clone N genes from seven human-infecting coronaviruses (SARS-CoV, MERS-CoV, HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, and SARS-CoV-2) into pUC57 vectors [25].
- Transform into E. coli DH5α cells and amplify plasmids using standard protocols.
- Extract plasmid DNA using commercial mini-prep kits (e.g., Omega Plasmid Mini Kit).
RT-qPCR Analysis:
- Prepare templates by diluting plasmids to 10^7 copies/μL in nuclease-free water.
- Perform RT-qPCR reactions using commercial master mixes with the following cycling conditions:
- Reverse transcription: 50°C for 15 minutes
- Initial denaturation: 95°C for 2 minutes
- 40 cycles of: 95°C for 15 seconds, 60°C for 1 minute
- Compare Ct values between structure-based primers and reference primers (e.g., US CDC primers).
Mismatch Tolerance Testing:
- Systematically introduce mutations at different positions in primers (first, second, and third nucleotides from 3' end, and middle positions).
- Test each mutated primer against the SARS-CoV-2 N gene template.
- Confirm that mutations at the 3' end (tryptophan codon) have the most significant impact on amplification efficiency.
Nested PCR Protocol for Enhanced Sensitivity
This protocol describes a conventional nested PCR approach targeting the N gene for detection of SARS-CoV-2 in clinical and animal samples.
Reagents and Equipment:
- RNA extraction kit (e.g., ISOLATE II RNA Mini Kit)
- Reverse transcription kit (e.g., SensiFAST cDNA Synthesis Kit)
- PCR master mix (e.g., My Taq HS Red Mix)
- Nested PCR primers targeting SARS-CoV-2 N gene:
- External forward: 5'-CGCATTGGCAATGTTGTTC-3'
- External reverse: 5'-TGGCACCTGTGTAGCGTAAC-3' (633bp product)
- Internal forward: 5'-GCTTCTGGGACCAATGGTA-3'
- Internal reverse: 5'-CAGGTAAGCGTAAAACTCATC-3' (248bp product) [17]
- Thermal cycler
- Gel electrophoresis system
Procedure:
RNA Extraction:
- Extract RNA from 200μL of clinical sample (nasopharyngeal swab, oropharyngeal swab) using commercial viral RNA extraction kits according to manufacturer's instructions.
- Elute RNA in 50μL of nuclease-free water.
- Quantify RNA concentration and purity using spectrophotometry (e.g., NanoDrop 2000).
Reverse Transcription:
- Prepare 20μL reaction mixture containing:
- 7μL extracted RNA
- 8μL DEPC-treated water
- 4μL TransAmp buffer
- 1μL reverse transcriptase enzyme
- Incubate at 25°C for 10 minutes, 42°C for 15 minutes, and 80°C for 5 minutes.
- Store cDNA at -20°C if not used immediately.
- Prepare 20μL reaction mixture containing:
First Round PCR:
- Prepare 25μL reaction mixture containing:
- 12.5μL PCR master mix
- 4μL cDNA template
- 1μL each external primer (10 pmol/μL)
- 6.5μL PCR-grade water
- Cycling conditions:
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of: 95°C for 30 seconds, 49°C for 30 seconds, 72°C for 45 seconds
- Final extension: 72°C for 5 minutes
- Prepare 25μL reaction mixture containing:
Second Round PCR:
- Dilute first-round PCR product 1:50 in PCR-grade water.
- Prepare 25μL reaction mixture containing:
- 12.5μL PCR master mix
- 0.5μL diluted first-round PCR product
- 1μL each internal primer (10 pmol/μL)
- 10μL PCR-grade water
- Cycling conditions:
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of: 95°C for 30 seconds, 51°C for 30 seconds, 72°C for 30 seconds
- Final extension: 72°C for 5 minutes
Detection and Analysis:
- Analyze 10μL of second-round PCR product by 2% agarose gel electrophoresis.
- Visualize bands using UV transillumination after ethidium bromide staining.
- Expected product size: 248bp.
- Include positive and negative controls in each run.
Troubleshooting:
- If non-specific amplification occurs, optimize annealing temperature (47-53°C for first round, 49-55°C for second round).
- If sensitivity is inadequate, increase number of cycles to maximum 40 per round.
- To prevent contamination, perform pre- and post-PCR procedures in separate areas.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for SARS-CoV-2 Primer Design and Validation
| Reagent/Kit | Manufacturer/Reference | Function | Application Notes |
|---|---|---|---|
| pUC57-N Plasmid Vectors | Beijing Genomics Institute [25] | Contains cloned N genes of human coronaviruses | Enables specificity testing against related coronaviruses |
| SensiFAST cDNA Synthesis Kit | Bioline, Meridian Bioscience [17] | Reverse transcription of viral RNA | High efficiency conversion of RNA to cDNA |
| My Taq HS Red Mix | Bioline, Meridian Bioscience [17] | PCR amplification for nested protocols | Contains all components except primers and template |
| MiniBEST Viral RNA/DNA Extraction Kit | TaKaRa [25] | Nucleic acid extraction from clinical samples | Suitable for both RNA and DNA extraction |
| CleanPlex SARS-CoV-2 Panel | Paragon Genomics [26] | Whole genome sequencing for validation | Identifies mutations in primer binding regions |
| QIAamp Viral RNA Mini Kit | Qiagen [27] [2] | RNA extraction from clinical specimens | Compatible with various sample types |
Strategic primer design targeting the N gene and ORF1ab region requires integration of structural biology principles, bioinformatic monitoring of viral evolution, and implementation of enhanced sensitivity methods like nested PCR. Structure-based approaches focusing on structurally constrained residues and AI-assisted primer discovery offer promising avenues for developing variant-resilient detection assays. The experimental protocols outlined provide robust frameworks for designing and validating such primer systems, with nested PCR formats delivering the sensitivity required for detecting low viral loads in clinical, surveillance, and animal samples. As SARS-CoV-2 continues to evolve, these advanced primer design strategies will remain essential for maintaining diagnostic accuracy in both human and animal populations.
Within the framework of advanced molecular diagnostics for SARS-CoV-2, the evolution of PCR methodologies from conventional assays to sophisticated nested formats represents a significant leap in detection capability. Reverse transcription real-time quantitative PCR (qRT-PCR) remains the gold standard for clinical detection of SARS-CoV-2 RNA [30]. However, its sensitivity limitations, with reported positive rates of throat swab samples varying from 30% to 60%, have prompted the development of more advanced detection strategies [30]. The one-step single-tube nested qRT-PCR (OSN-qRT-PCR) has emerged as a powerful alternative, demonstrating superior sensitivity for detecting SARS-CoV-2 in patients with low viral loads [30]. This protocol details both conventional and OSN-qRT-PCR procedures, providing researchers with optimized methodologies for SARS-CoV-2 detection across various sample types and viral load scenarios.
Principle and Advantages of OSN-qRT-PCR
Nested PCR traditionally employs two sequential amplification reactions, each using a different pair of primers, leading to significant increases in both sensitivity and specificity [30]. The conventional approach requires physically transferring the first-round amplification product to a new tube for the second reaction, which is not only laborious but also increases the risk of laboratory contamination [31].
The OSN-qRT-PCR format innovates by containing both amplification reactions within a single tube. This is achieved through primer engineering and thermal cycling optimization, where external primers function at a higher annealing temperature than the internal primers, with switching between the two PCR stages controlled by precise temperature changes [31]. This single-tube approach maintains the sensitivity benefits of nested PCR while reducing contamination risks and streamlining the workflow [31] [30].
For SARS-CoV-2 detection, OSN-qRT-PCR has demonstrated remarkable clinical performance, detecting the virus at concentrations as low as 189.1 copies/mL for the N gene and 194.74 copies/mL for ORF1ab [30]. In direct comparisons, OSN-qRT-PCR has shown positive detection rates of 82.35% compared to 58.82% for conventional qRT-PCR in clinical samples from COVID-19 patients [30].
Experimental Protocols
Sample Preparation and RNA Extraction
Wastewater Sample Concentration
For environmental surveillance using wastewater samples, effective concentration methods are critical:
- Employ polyethylene glycol (PEG) precipitation for viral concentration due to its ease of use and minimal equipment requirements [31] [32].
- Alternative concentration methods include ultracentrifugation, flocculation, and filtration, though performance varies based on experimental conditions [31].
RNA Extraction
- Extract total RNA using chaotropic RNA extraction methods or magnetic beads-based systems [32].
- For clinical samples (throat swabs, nasopharyngeal swabs, sputum, blood), use commercial RNA extraction kits following manufacturer protocols [30] [33].
- Elute RNA in 50-80 μL of elution buffer.
- Quantify and assess RNA purity using spectrophotometry (NanoDrop) [1].
Primer and Probe Design
Conventional qRT-PCR Assays
- Target genes: Focus on conserved regions of SARS-CoV-2 genome, including nucleocapsid phosphoprotein (N), spike protein (S), membrane (M), ORF1ab, and RNA-dependent RNA polymerase (RdRp) genes [31] [34].
- N gene assays: Demonstrate high efficiency with LoD of 20-80 copies/μL for different regions (N1, N2) [33].
- M gene assays: Provide an alternative target with high conservation and lower mutation rate, achieving LoD of 100 copies/mL [34].
- Design primers and probes using software such as Primer3Plus with the following characteristics:
- Primer length: 18-25 bases
- Tm: 55-65°C
- Amplicon size: 65-100 bp for optimal qRT-PCR efficiency
- Validate specificity using BLAST against the human genome and other coronaviruses.
Table 1: Primer and Probe Sequences for SARS-CoV-2 Detection
| Target | Primer/Probe Name | Sequence (5'→3') | Amplicon Size | Reference |
|---|---|---|---|---|
| S gene | HOTSpikeFw | AGTGCAAATTGTAGAGGTTGATC | 88 bp | [31] |
| HOTSpikeRv | TCTGATTTCTGCAGCTCTAATTA | |||
| P-LANL_4.1 | FAM-GGCAGACTTCAAAGTTTGCA-BHQ1 | |||
| N gene | N3-F | GGGAGCCTTGAATACACCAAAA | 72 bp | [31] |
| N3-R | TGTAGAGCAGCGTTGTTGGA | |||
| N3-P | FAM-ACCCGCATTACGTTTGGTGGACC-BHQ1 | |||
| N gene (nested external) | Ext2019nCorVF | GGCAGTAACCAGAATGGAGA | 335 bp | [1] |
| Ext2019nCorVR | CTCAGTTGCAACCCATATGAT | |||
| N gene (nested internal) | intF | CACCGCTCTCACTCAACAT | 212 bp | [1] |
| intR | CATAGGGAAGTCCAGCTTCT |
OSN-qRT-PCR Assays
- Design two primer pairs: external primers that amplify a larger fragment (150-300 bp) and internal primers that bind within the first amplicon, producing a smaller fragment (80-150 bp) [31].
- Ensure a significant Tm difference between external and internal primers (3-5°C) to facilitate thermal cycling optimization.
- Utilize a single TaqMan probe that binds to the region amplified by both primer sets.
- For SARS-CoV-2 S gene OSN-qRT-PCR:
- External primers: LANLMay4.1Fw and LANLMay4.1Rv (155 bp product)
- Internal primers: InnerSpikeFw and InnerSpikeRv (85 bp product)
- Common probe: P-LANL_4.1 [31]
Conventional qRT-PCR Protocol
Reaction Setup
- Prepare reaction mix using commercial one-step RT-PCR kits (e.g., SuperScript III One-Step RT-PCR System) [33].
- Standard 20 μL reaction:
- 2× Master Mix: 10 μL
- Platinum Enzyme Mix: 0.4 μL
- Forward primer (10 μM): 0.75 μL
- Reverse primer (10 μM): 0.75 μL
- Probe (10 μM): 0.75 μL
- ROX reference dye (optional): 0.4 μL
- RNA template: 5 μL
- Nuclease-free water: 2.7 μL
- Cost-effective half-reaction (10 μL total):
- 2× Master Mix: 5 μL
- Platinum Enzyme Mix: 0.2 μL
- Forward primer (10 μM): 0.375 μL
- Reverse primer (10 μM): 0.375 μL
- Probe (10 μM): 0.375 μL
- ROX reference dye (optional): 0.2 μL
- RNA template: 4 μL
- Nuclease-free water: 1.475 μL [33]
Thermal Cycling Conditions
- Reverse transcription: 50°C for 15-30 minutes
- Initial denaturation: 95°C for 2 minutes
- Amplification (40-45 cycles):
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 55-60°C for 30-35 seconds
- Data collection during annealing/extension phase [33] [35].
One-Step Single-Tube Nested qRT-PCR Protocol
Reaction Setup
- Prepare reaction mix similarly to conventional qRT-PCR but include both primer sets:
- 2× Master Mix: 10 μL
- Reverse transcriptase: 1 μL
- External forward primer (10 μM): 0.5 μL
- External reverse primer (10 μM): 0.5 μL
- Internal forward primer (10 μM): 0.5 μL
- Internal reverse primer (10 μM): 0.5 μL
- Probe (10 μM): 0.75 μL
- RNA template: 5 μL
- Nuclease-free water: 1.75 μL
- Total reaction volume: 20 μL [31] [30]
Optimized Thermal Cycling Conditions
- Reverse transcription: 50°C for 15-30 minutes
- Initial denaturation: 95°C for 2 minutes
- First-stage amplification (10-15 cycles):
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 65-68°C (higher Tm for external primers) for 30 seconds
- Second-stage amplification (35-40 cycles):
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 55-60°C (lower Tm for internal primers) for 30 seconds
- Data collection during the second-stage annealing/extension phase [31] [30].
Diagram 1: Comparative Workflow of Conventional and OSN-qRT-PCR Methods
Performance Comparison and Validation
Analytical Sensitivity and Detection Limits
Table 2: Analytical Performance Comparison of SARS-CoV-2 Detection Methods
| Method | Target Gene | Limit of Detection (copies/mL) | Positive Detection Rate | Applications | Reference |
|---|---|---|---|---|---|
| Conventional qRT-PCR | ORF1ab | 520.1 (95% CI: 363.23-1145.69) | 58.82% (20/34 clinical samples) | Clinical diagnosis, wastewater surveillance | [30] |
| N | 528.1 (95% CI: 347.7-1248.7) | ||||
| N1 | 20 copies/μL | - | Clinical diagnosis | [33] | |
| N2 | 80 copies/μL | - | Clinical diagnosis | [33] | |
| OSN-qRT-PCR | ORF1ab | 194.74 (95% CI: 139.7-430.9) | 82.35% (28/34 clinical samples) | Low viral load samples, wastewater surveillance | [30] |
| N | 189.1 (95% CI: 130.9-433.9) | ||||
| Nested PCR (Two-Step) | N | ~50 copies/μL (Ct ~31.5) | 95% sensitivity, 100% specificity | Animal samples, low viral loads | [4] |
| ddPCR | ORF1ab | 401.8 (95% CI: 284.8-938.3) | 67.65% (23/34 clinical samples) | Research, quantification | [30] |
| N | 336.8 (95% CI: 244.6-792.5) | ||||
| M gene qRT-PCR | M | 100 copies/mL | Comparable to commercial kits | Variant detection, clinical diagnosis | [34] |
Assay Validation and Quality Control
Positive Controls
- Use encapsidated RNA mimic systems (ENRM for N gene, ESRM for S gene) as reliable positive controls that simulate viral particles [31] [32].
- Alternatively, use SARS-CoV-2 pseudovirus RNA or armored RNA at known concentrations [30] [36].
- Include synthetic RNA transcripts for standard curve generation and efficiency calculations [35].
Specificity Testing
- Validate assay specificity against other human coronaviruses (HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1) and respiratory pathogens.
- For animal samples, test against related animal coronaviruses (canine coronavirus, feline infectious peritonitis virus) [4].
- Include no-template controls (NTC) and negative sample controls in each run.
Efficiency Calculations
- Perform serial dilutions of standard RNA (10^5 to 10^1 copies/μL) in triplicate.
- Generate standard curves by plotting Ct values against log10 RNA concentration.
- Calculate amplification efficiency using the formula: E = [10^(-1/slope) - 1] × 100%
- Acceptable efficiency: 90-110% with R² > 0.95 [33] [35].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for SARS-CoV-2 PCR Detection
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| RNA Extraction Kits | ISOLATE II RNA Mini Kit, Abbott mSample Preparation System | Nucleic acid purification from clinical/environmental samples | Ensure compatibility with sample type; evaluate yield and purity |
| One-Step RT-PCR Master Mixes | SuperScript III One-Step RT-PCR System, Luna Universal Probe One-Step RT-PCR Kit | Combined reverse transcription and PCR amplification | Optimize enzyme concentrations; check compatibility with fast cycling |
| Positive Controls | Encapsidated RNA Mimics (ENRM/ESRM), SARS-CoV-2 pseudovirus, armored RNA | Assay validation, process control, standard curve generation | Mimics should resemble target virus structure; use at predetermined concentrations |
| Primers/Probes | CDC N1/N2 assays, Charité E gene assay, custom OSN primers | Target-specific amplification | Validate specificity; check for mutations in primer binding sites |
| Inhibition Controls | RNAse P, exogenous internal controls (ASBVd) | Detection of PCR inhibitors in sample extracts | Should be spiked into lysis buffer to control for entire extraction process |
| Quantification Standards | Synthetic RNA transcripts, digital PCR quantified standards | Absolute quantification, assay calibration | Use serially diluted standards for efficiency calculations |
Applications and Implementation Guidelines
Clinical Sample Analysis
- For diagnostic laboratories with high sample throughput, conventional qRT-PCR remains the standard approach due to established protocols and regulatory approvals.
- Implement OSN-qRT-PCR for cases with low viral loads or discrepant results, particularly in immunocompromised patients or convalescent testing.
- Use half-reactions (10 μL total volume) to reduce costs during mass testing while maintaining sensitivity [33].
Wastewater-Based Epidemiology
- Apply PEG precipitation combined with OSN-qRT-PCR for optimal detection of SARS-CoV-2 in wastewater with low viral loads [31] [32].
- Target multiple genes (N and S) to enhance detection reliability and track variants.
- Correlate wastewater viral levels with clinical case numbers, considering 3-5 day temporal offsets for optimal correlation [31].
Animal Surveillance
- Implement nested PCR formats for large-scale animal sampling due to lower costs compared to real-time PCR [4].
- Target N gene for its higher conservation and stability across species [4].
- Establish specific protocols for different sample types (oropharyngeal, conjunctival, nasal swabs) from susceptible animals.
Troubleshooting and Optimization
Common Issues and Solutions
- Low sensitivity: Check RNA quality, increase template volume, switch to OSN-qRT-PCR format, or optimize primer concentrations.
- Inconsistent replicates: Verify pipetting accuracy, ensure thorough mixing of reagents, and check thermal cycler calibration.
- High background noise: Optimize probe concentration, check for probe degradation, or increase annealing temperature.
- Inhibition issues: Dilute samples, add inhibition removal steps, or use internal controls to detect inhibition.
Assay Selection Guidelines
- High viral loads: Conventional qRT-PCR provides cost-effective, reliable results.
- Low viral loads (<500 copies/mL): OSN-qRT-PCR or digital PCR offer superior detection capabilities.
- Variant surveillance: Multi-target assays (ORF1ab, S, N) or M gene-targeted approaches reduce false negatives due to mutations.
- Resource-limited settings: Nested PCR (two-step or OSN) provides high sensitivity without requiring real-time PCR instrumentation.
The continuous evolution of SARS-CoV-2 demands flexible molecular detection strategies. While conventional qRT-PCR remains the workhorse for diagnostic laboratories, OSN-qRT-PCR and related nested approaches offer enhanced sensitivity for challenging samples and specialized applications. The protocols detailed herein provide researchers with robust methodologies adaptable to various surveillance and diagnostic scenarios.
The accuracy of SARS-CoV-2 detection, particularly when using highly sensitive methods like nested PCR, is fundamentally dependent on the upstream process of viral RNA extraction. The efficiency of this recovery process varies significantly across different sample matrices, such as nasopharyngeal (NP) swabs, sputum, and wastewater, each presenting unique compositional challenges that can inhibit downstream molecular analysis [37] [38]. This application note provides a detailed, comparative guide to optimized RNA extraction protocols from these distinct sample types, contextualized within a research framework aimed at developing a robust nested PCR assay for SARS-CoV-2. We summarize critical performance data and provide step-by-step methodologies to enable researchers to maximize viral RNA yield and purity, thereby ensuring the reliability of their detection assays.
Comparative Performance of RNA Extraction Methods
The selection of an RNA extraction method is a critical determinant for the success of downstream nested PCR. The following table summarizes key performance characteristics of various approaches as validated for different sample types.
Table 1: Performance Comparison of RNA Extraction Methods for SARS-CoV-2 Detection
| Sample Type | Extraction Method / Principle | Key Performance Characteristics | Reference |
|---|---|---|---|
| Wastewater | NS2 Protocol: Neutral phenol-chloroform + magnetic silica beads (NucliSENSⓇ kit) + final column purification | • Significantly higher SARS-CoV-2 RNA detection than WW-designed silica column protocol (Z) (p < 0.0001)• Complete removal of RT-qPCR inhibitors [37] | [37] |
| Wastewater | Silica Beads (SHIFT-SP): Magnetic silica bead-based, optimized low-pH binding and tip-based mixing | • Rapid (6-7 min) and high-yield (extracts nearly all NA in sample)• High yield achieved with low pH (4.1) and tip-based mixing for 1-2 min [39] | [39] |
| Wastewater | Zymo Quick RNA Viral Kit (Lysis buffer principle) | • Outperformed other kits (lysis and bead-beating) in viral RNA yield and cost-effectiveness for wastewater [40] | [40] |
| Wastewater | Promega Total Nucleic Acid Kit (Silica column) | • Used for extraction from large-volume (40 mL) protease-treated wastewater samples [41] | [41] |
| Wastewater | Bead-beating (Vigorous mechanical disruption) | • Significantly increased RNA yield, with efficiency reaching up to 82.18% [38] | [38] |
| Sputum | Automated System (Zhijiang kit on EX3600 platform) | • RNA eluted in 50 µL for subsequent reverse transcription and nested PCR [24] | [24] |
| NP/Throat Swabs | ISOLATE II RNA Mini Kit (Silica column) | • Validated for use in nested PCR; showed 100% sensitivity and specificity [17] | [17] |
Detailed Experimental Protocols
Protocol A: Comprehensive RNA Extraction from Wastewater using the NS2 Method
This protocol, adapted from a comparative study, is designed for maximum recovery and inhibitor removal from complex wastewater matrices [37].
Materials & Reagents:
- NucliSENSⓇ easyMAGⓇ Lysis Buffer
- Phenol-chloroform-isoamyl alcohol (25:24:1, pH 7.8–8.2)
- NucliSENSⓇ easyMAGⓇ Magnetic Silica
- Wash Buffers 1, 2, and 3 (e.g., from NucliSENSⓇ kit)
- OneStep PCR Inhibitor Removal Kit (ZymoResearch)
- Separation tube with silicon grease/SiO2 mixture (90:10 w/w)
Procedure:
- Sample Lysis:
- Add 10 mL of NucliSENSⓇ lysis buffer to 5 mL of raw wastewater sample.
- Homogenize by vortexing at high speed for 1 minute.
- Incubate the mixture at room temperature for 10 minutes. This mixture is now the lysate.
Neutral Phenol-Chloroform Treatment:
- In a chemical fume hood, add an equal volume of neutral phenol-chloroform-isoamyl alcohol to the lysate.
- Mix thoroughly and centrifuge using a swinging bucket rotor to separate the aqueous and organic phases.
- Carefully transfer the upper aqueous phase containing the RNA to a new tube.
RNA Capture with Magnetic Silica:
- Combine the aqueous phase with magnetic silica particles and a binding buffer.
- Incubate with continuous mixing to allow RNA to bind to the silica.
Washing:
- Pellet the silica particles using a magnetic rack and discard the supernatant.
- Wash the beads sequentially with Wash Buffer 1, Wash Buffer 2, and an alcohol-based Wash Buffer 3 to remove impurities and inhibitors.
Final Purification & Elution:
- For maximum purity, perform a final purification step using the OneStep PCR Inhibitor Removal resin column.
- Elute the purified RNA in 50 µL of nuclease-free water.
Protocol B: High-Speed RNA Extraction using SHIFT-SP for Various Samples
This optimized magnetic bead protocol is suitable for high-throughput applications and can be adapted for different sample types [39].
Materials & Reagents:
- Lysis Binding Buffer (LBB) at pH 4.1 (containing chaotropic salts)
- Silica-coated magnetic beads
- Wash Buffer (e.g., alcohol-based)
- Low-EDTA TE Buffer or Nuclease-free Water (for elution)
Procedure:
- Sample Lysis and Binding:
- Mix the sample (e.g., swab medium, processed sputum, or concentrated wastewater) with LBB (pH 4.1) to denature proteins and release nucleic acids.
- Add 30-50 µL of magnetic silica beads.
- For binding, use a tip-based mixing method: repeatedly aspirate and dispense the binding mix for 1-2 minutes. This exposes the beads to the entire sample more effectively than orbital shaking, significantly improving binding efficiency and speed.
Washing:
- Capture beads on a magnetic stand and discard the supernatant.
- Wash the beads twice with an appropriate Wash Buffer to remove salts and other contaminants.
Elution:
- Elute the pure RNA in a small volume (e.g., 20-50 µL) of pre-warmed (60-70°C) low-EDTA TE Buffer or nuclease-free water. Heating during elution increases yield.
Protocol C: Routine RNA Extraction from NP Swabs and Sputum for Nested PCR
This is a standardized column-based protocol for clinical samples like swabs and sputum [24] [17].
Materials & Reagents:
- ISOLATE II RNA Mini Kit (Bioline) or equivalent silica-column kit
- SensiFAST cDNA Synthesis Kit (Bioline)
- Ethanol (96-100%)
- Proteinase K (optional, for sputum pre-treatment)
Procedure:
- Sample Pre-treatment:
- NP/Throat Swabs: Vortex the swab in transport medium. For sputum, liquefy it using a reducing agent or proteinase K, then centrifuge to remove debris [24].
- Begin with 200 µL of the liquid sample.
Lysis:
- Mix the sample with a lysis buffer containing a chaotropic salt (e.g., guanidine thiocyanate) to inactivate nucleases and release RNA.
Binding:
- Add ethanol to the lysate and apply the entire volume to a silica column. Centrifuge to bind the RNA to the membrane.
Washing:
- Wash the column twice with provided wash buffers to remove contaminants.
Elution:
- Elute the RNA in 50 µL of nuclease-free water.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for RNA Extraction and Their Functions
| Reagent / Kit | Primary Function | Application Notes |
|---|---|---|
| NucliSENSⓇ kit | Magnetic silica-based extraction | Optimal for difficult matrices like wastewater; often used with a phenol-chloroform pre-step (NS2 protocol) [37] |
| Phenol-Chloroform | Organic extraction and phase separation | Effectively removes proteins, lipids, and PCR inhibitors; requires careful handling in a fume hood [37] |
| Lysis Binding Buffer (Low pH) | Creates conditions for NA binding to silica | A pH of ~4.1 reduces electrostatic repulsion between silica and RNA, dramatically improving binding efficiency [39] |
| Magnetic Silica Beads | Solid-phase matrix for NA binding | Enable automation and rapid processing; performance depends on mixing efficiency (e.g., tip-based mixing) [37] [39] |
| Silica Columns | Solid-phase matrix for NA binding | Simple and effective for clinical samples; can clog with overly concentrated or large-volume wastewater samples [37] |
| OneStep PCR Inhibitor Removal Kit | Final purification step | Removes residual humic acids, metals, and other inhibitors common in wastewater, improving PCR reliability [37] |
| Promega Total Nucleic Acid Kit | Simultaneous extraction of RNA and DNA | Suitable for wastewater surveillance where co-detection of other pathogens or fecal normalization (e.g., PMMoV) is desired [41] [42] |
Integrated Workflow for Sample Processing and Analysis
The following diagram illustrates the complete pathway for processing different sample types, from collection to the final nested PCR result, integrating the protocols described above.
The successful detection of SARS-CoV-2 via nested PCR is contingent upon a sample-specific and optimized RNA extraction strategy. For NP swabs and sputum, standardized silica-column protocols provide a robust balance of ease and performance. In contrast, the complex and inhibitor-rich nature of wastewater demands more rigorous methods, such as the NS2 protocol combining organic extraction with magnetic silica purification, or the rapid and highly efficient SHIFT-SP method. The protocols and data summarized in this application note provide a foundation for researchers to build reliable and sensitive workflows for SARS-CoV-2 detection across diverse sample types, directly supporting the development and validation of novel nested PCR assays.
The COVID-19 pandemic has highlighted the critical importance of understanding zoonotic disease dynamics, particularly the transmission of SARS-CoV-2 between humans and animals. Among companion animals, cats (Felis catus) demonstrate high susceptibility to SARS-CoV-2 infection due to their angiotensin-converting enzyme 2 (ACE2) receptor similarity to humans [43] [44]. Reverse zoonosis (human-to-animal transmission) has been documented globally, with studies reporting seroprevalence rates of up to 30.3% in cats from COVID-19-positive households [44]. Molecular surveillance of SARS-CoV-2 in animal populations is therefore essential for a comprehensive One Health approach to pandemic management.
Conventional real-time reverse transcription PCR (RT-qPCR) remains the gold standard for SARS-CoV-2 detection but presents limitations for large-scale animal surveillance due to substantial equipment costs and reagent expenses [4]. This application note details the development, validation, and implementation of a conventional nested PCR assay targeting the SARS-CoV-2 N gene, providing researchers with a highly sensitive, specific, and cost-effective alternative for detecting active SARS-CoV-2 infection in cats [17] [1].
Assay Validation and Performance Characteristics
The nested PCR assay for SARS-CoV-2 detection has undergone rigorous validation using both human and feline samples, demonstrating excellent performance characteristics for zoonotic research applications.
Table 1: Performance Metrics of Nested PCR for SARS-CoV-2 Detection
| Parameter | Performance | Experimental Conditions |
|---|---|---|
| Analytical Sensitivity | ||
| RNA Detection Limit | 0.015 ng/μL | Extracted RNA from SARS-CoV-2 reference strain [17] |
| Viral Copy Detection Limit | ~50 copies/μL | Corresponding to Ct value of 31.5 [4] [10] |
| Diagnostic Performance | ||
| Sensitivity | 95-100% | Compared to RT-qPCR and LAMP assays [17] [4] |
| Specificity | 100% | No cross-reactivity with CCV, FIPV, FCV, FHV [17] [4] |
| Assay Agreement | ||
| Kappa Value | 0.829 (Excellent) | Comparison with RT-qPCR reference method [4] |
The assay demonstrates particular effectiveness in detecting low viral loads, with positive detection reported in samples with Ct values as high as 31.5 [4]. This sensitivity is crucial for surveillance studies, as infected cats often present with asymptomatic infections or low viral loads [43]. The primer sets targeting the N gene have shown no cross-reactivity with other common feline pathogens, including feline coronavirus (FCoV), feline calicivirus (FCV), feline herpesvirus (FHV), Chlamydia, and Mycoplasma spp., ensuring reliable results in field conditions [17].
Experimental Protocols
Primer Design and Specifications
The nested PCR assay employs two primer pairs targeting conserved regions of the SARS-CoV-2 nucleocapsid (N) gene, designed based on the reference sequence MN908947.3 (isolate Wuhan-Hu-1) [17] [1].
Table 2: Primer Sequences for SARS-CoV-2 N Gene Amplification
| Primer Name | Sequence (5' → 3') | Genome Position | Amplicon Size | Role |
|---|---|---|---|---|
| Ext2019nCorVF | GGCAGTAACCAGAATGGAGA |
28346-28365 | 335 bp | External Forward |
| Ext2019nCorVR | CTCAGTTGCAACCCATATGAT |
28681-28661 | 335 bp | External Reverse |
| intF | CACCGCTCTCACTCAACAT |
28432-28450 | 212 bp | Internal Forward |
| intR | CATAGGGAAGTCCAGCTTCT |
28643-28624 | 212 bp | Internal Reverse |
Sample Collection and RNA Extraction
Sample Collection: Collect nasopharyngeal, oropharyngeal, and conjunctival swabs from cats using standard veterinary techniques. Place swabs immediately into virus transport media and store at 4°C for short-term storage (up to 24 hours) or -80°C for long-term preservation [17] [43].
RNA Extraction: Extract total RNA using commercial kits such as the ISOLATE II RNA Mini Kit (Bioline). Use approximately 200 μL of transport media for extraction, following manufacturer's instructions. Elute RNA in 50-100 μL of nuclease-free water [17].
RNA Quantification and Quality Control: Measure RNA concentration and purity using spectrophotometry (e.g., NanoDrop 2000). Acceptable samples should have A260/A280 ratios between 1.8 and 2.1. Store extracted RNA at -80°C if not used immediately [17].
Reverse Transcription and Nested PCR Amplification
Reverse Transcription
- Prepare 20 μL reaction mixture containing:
- 7 μL extracted RNA
- 8 μL DEPC-treated water
- 4 μL TransAmp buffer
- 1 μL reverse transcriptase enzyme
- Use the following thermal cycling conditions:
- 25°C for 10 minutes
- 42°C for 15 minutes
- 80°C for 5 minutes
- Store synthesized cDNA at -20°C for immediate use or -80°C for long-term storage [17].
First Round PCR
- Prepare 25 μL reaction mixture:
- 12.5 μL My Taq HS Red Mix
- 4 μL cDNA template
- 1 μL each external primer (10 pmol/μL)
- 6.5 μL PCR-grade water
- Use the following amplification protocol:
- Initial denaturation: 95°C for 1 minute
- 35 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing: 54.6°C for 15 seconds
- Extension: 72°C for 10 seconds
- Final extension: 72°C for 1 minute [17]
Second Round PCR
- Prepare 25 μL reaction mixture:
- 12.5 μL My Taq HS Red Mix
- 0.5 μL first-round PCR product
- 1 μL each internal primer (10 pmol/μL)
- 10 μL PCR-grade water
- Use the same amplification protocol as the first round [17].
Product Analysis and Confirmation
Gel Electrophoresis:
- Prepare 2% agarose gel in 1× TAE buffer with ethidium bromide (10 ng/mL)
- Load 5-10 μL of second-round PCR product alongside appropriate DNA ladder
- Run gel at 120-150 V for 30 minutes
- Visualize under UV transilluminator
- Positive samples show clear band at 212 bp [17]
Sequencing Confirmation (Optional):
- Purify PCR products using commercial kits (e.g., Exo-CIP Rapid PCR Cleanup Kit)
- Perform Sanger sequencing using internal primers
- Analyze sequences using BLAST against SARS-CoV-2 reference databases [17]
The Scientist's Toolkit
Table 3: Essential Research Reagents and Equipment
| Category | Specific Product/Kit | Application/Function |
|---|---|---|
| RNA Extraction | ISOLATE II RNA Mini Kit (Bioline) | Total RNA isolation from swab samples |
| cDNA Synthesis | SensiFAST cDNA Synthesis Kit (Bioline) | Reverse transcription of viral RNA to cDNA |
| PCR Master Mix | My Taq HS Red Mix (Bioline) | Ready-to-use PCR mix with loading dye |
| Positive Control | Inactivated SARS-CoV-2 USA-WA1/2020 | Assay performance verification |
| Primer Specificity Controls | FCoV, FCV, FHV positive samples | Verification of assay specificity |
| Thermal Cyclers | QB96, SaCycler-96, FluoroCycler | DNA amplification |
| Electrophoresis | Agarose gel system with UV transilluminator | PCR product visualization and analysis |
| Sequencing | Big Dye Terminator v3.1 Kit, ABI 3500xl | Amplicon confirmation and variant identification |
Application in Feline SARS-CoV-2 Surveillance
The implemented nested PCR assay has successfully detected natural SARS-CoV-2 infections in feline populations across multiple countries:
- Bulgaria: During the first COVID-19 wave, six cats with respiratory symptoms tested positive for SARS-CoV-2 using this nested PCR assay, with confirmation by sequence analysis [17].
- China: A surveillance study of 458 cats in 2023-2024 detected SARS-CoV-2 in 1.5% of samples, identifying the Omicron variant BA.5.2 in all positive cases [43].
- Argentina: Validation studies demonstrated 95% sensitivity and 100% specificity when testing 140 dog and cat samples, effectively detecting infections with low viral loads [4].
- Egypt: Research confirmed the transmission of the Delta variant from humans to domestic cats, with whole-genome sequencing revealing 99.6-99.7% nucleotide identity to human SARS-CoV-2 viruses [45].
Most infected cats present with mild respiratory symptoms or remain asymptomatic, though some may show respiratory distress or sneezing [43]. Co-infections with other feline pathogens like feline calicivirus have been documented, highlighting the importance of specific SARS-CoV-2 detection methods [43].
Workflow and Technical Diagrams
Nested PCR Workflow for SARS-CoV-2 Detection in Cats
Nested PCR Primer Binding Strategy
The conventional nested PCR assay presented here provides researchers with a reliable, sensitive, and cost-effective method for detecting active SARS-CoV-2 infection in cats. Its high sensitivity (95-100%) and specificity (100%), coupled with minimal equipment requirements, make it particularly suitable for large-scale surveillance studies and laboratories with limited resources. The assay's proven effectiveness across multiple countries and variants underscores its value in ongoing One Health initiatives aimed at monitoring SARS-CoV-2 at the human-animal interface. Implementation of this protocol will enhance our understanding of reverse zoonotic transmission dynamics and contribute to early warning systems for potential spillback events.
Wastewater-Based Epidemiology (WBE) has emerged as a powerful public health tool for monitoring infectious disease agents, including SARS-CoV-2, at the population level. This approach involves the systematic collection and analysis of wastewater to identify and quantify pathogens circulating in a community. The COVID-19 pandemic has accelerated the development and standardization of WBE methods, transforming it from an "obscure, underused scientific tool to a rigorous, widely accepted approach for comprehensive disease monitoring" [46]. WBE offers a unique advantage over clinical testing by capturing data from both symptomatic and asymptomatic individuals, providing public health officials with real-time, cost-effective insights into community transmission dynamics days before clinical cases are reported [47].
The application of WBE to SARS-CoV-2 surveillance is particularly valuable because infected individuals shed viral RNA in feces, saliva, sputum, and mucus, which ultimately reaches wastewater systems [47]. This review focuses on the integration of highly sensitive molecular detection methods, specifically nested PCR assays, within WBE frameworks to enhance SARS-CoV-2 surveillance capabilities. By leveraging these advanced techniques, researchers can achieve the sensitivity necessary to detect low concentrations of viral RNA in complex wastewater matrices, enabling earlier outbreak detection and more informed public health responses.
Technical Background
Principles of Wastewater-Based Epidemiology
Wastewater-based epidemiology has evolved significantly since its early applications in the 1940s when U.S. epidemiologists first used wastewater to track polio outbreaks [47]. The fundamental premise of WBE is that wastewater systems aggregate biological materials from large populations, creating a composite sample that reflects the health status of the contributing community. For SARS-CoV-2 surveillance, this approach enables monitoring of entire communities regardless of testing availability, healthcare-seeking behaviors, or symptom status.
The timeline of WBE development demonstrates its expanding applications:
- 1940s: Initial use for polio surveillance in the United States [47]
- 1954: Application for schistosome studies in Brazil [47]
- 1980s: Monitoring for hepatitis using molecular techniques [47]
- 2005: Detection of illicit drugs in Italy's River Po [47]
- 2013: Early warning surveillance for norovirus, hepatitis A, and poliovirus in Israel [47]
- 2020: Rapid implementation for SARS-CoV-2 monitoring globally [47]
Nested PCR Technology
Nested polymerase chain reaction (nested PCR) is a modified amplification technique developed to enhance both the sensitivity and specificity of conventional PCR [48]. This method employs two successive amplification rounds with two distinct primer sets. The first round uses an outer primer pair to amplify the target sequence, while the second round uses an inner primer pair that binds internal to the first amplicon [48].
The key advantages of nested PCR include:
- Enhanced Sensitivity: The high total cycle number enables detection of extremely low template concentrations [48]
- Improved Specificity: The requirement for two independent primer binding events reduces non-specific amplification [48]
- Reliable Detection: Capable of amplifying almost any cDNA from minimal starting material [48]
A significant innovation in this field is the one-tube nested quantitative real-time PCR (qPCR), which addresses the major limitation of traditional nested PCR: contamination risk during transfer between amplification rounds [49]. This closed-tube approach maintains the sensitivity benefits while reducing procedural contamination, making it particularly valuable for diagnostic applications and wastewater surveillance [49].
Table 1: Comparison of PCR-Based Detection Methods
| Method | Principle | Sensitivity | Key Applications |
|---|---|---|---|
| Conventional qPCR | Single amplification with fluorescent probe detection | Moderate | High viral load detection, clinical diagnostics |
| Traditional Nested PCR | Two sequential amplifications with different primer sets | High (100-fold increase over conventional) | Low-abundance targets, complex samples |
| One-Tube Nested qPCR | Two amplifications in sealed tube with sequential primers | Very High (100 fg/μL demonstrated) | Low-load samples, wastewater surveillance |
| Multiplex PCR | Multiple primer sets for simultaneous target detection | Variable | Pathogen panels, variant screening |
Application to SARS-CoV-2 Surveillance
Wastewater Surveillance of SARS-CoV-2
The application of WBE to SARS-CoV-2 monitoring has expanded rapidly, with a recent scoping review identifying implementation in 46 countries across six continents during the first three years of the pandemic [50]. The geographical distribution of these studies reveals concentrated efforts in North America (34%), Europe (30.8%), and Asia (19.5%), with growing adoption worldwide [50]. This global implementation underscores the recognition of WBE as a valuable complement to clinical surveillance systems.
The primary applications of SARS-CoV-2 WBE include:
- Trend Analysis: Correlation of SARS-CoV-2 RNA trends with epidemiological data (40.7% of studies) [50]
- Variant Tracking: Monitoring the emergence and circulation of Variants of Concern (VOCs) [51]
- Early Warning: Detection of viral recrudescence 4-10 days before clinical case reporting [47]
- Coverage of Asymptomatic Cases: Capturing data from the estimated 80% of infected individuals who are asymptomatic or minimally symptomatic [47]
Nested PCR Enhancement for Wastewater Detection
The complex composition of wastewater presents significant challenges for pathogen detection, including the presence of reaction inhibitors and low target concentrations [51]. These factors necessitate highly sensitive detection methods like nested PCR to achieve reliable monitoring.
Research by Costa et al. (2025) demonstrated that nested PCR followed by next-generation sequencing (NGS) of small amplicons of the S gene significantly improved SARS-CoV-2 variant detection in wastewater [51]. Their approach enabled:
- Identification and frequency calculation of 29 mutations corresponding to Alpha, Beta, Gamma, Delta, Omicron, and P.2 variants
- Detection of Omicron-matching mutations before the lineage was officially classified as a Variant of Concern
- Cluster analysis showing wastewater sequences matched variants circulating in clinical populations [51]
The exceptional sensitivity of nested PCR is particularly valuable for early warning systems, as it can detect viral signals when community prevalence remains low. Furthermore, the method's robustness against inhibitors common in wastewater matrices enhances its utility for routine surveillance programs.
Table 2: Performance Comparison of SARS-CoV-2 Detection Methods in Wastewater
| Method | Detection Limit | Variant Discrimination | Implementation Complexity | Best Use Cases |
|---|---|---|---|---|
| RT-qPCR | Moderate | Limited (requires multiple assays) | Low | Routine trend monitoring, high prevalence |
| Digital PCR | High | Limited | Medium | Absolute quantification, low prevalence |
| Nested PCR + NGS | Very High | Comprehensive (mutation identification) | High | Variant tracking, early emergence |
| One-Tube Nested qPCR | Very High (100 fg/μL) | Moderate (with multiple probes) | Medium | Low viral load, resource-limited settings |
Experimental Protocols
Sample Collection and Processing
Proper sample collection and processing are critical for successful wastewater surveillance. The following protocol has been standardized and validated through the WastewaterSCAN program and similar initiatives [46].
Wastewater Sample Collection
- Collection Method: Collect 24-hour composite samples from wastewater treatment plant influent using automated refrigerated samplers
- Sample Volume: 200-500 mL of primary settled solids or 1L of raw wastewater
- Preservation: Store at 4°C during collection and process within 24 hours
- Transport: Maintain cold chain (4°C) during transport to laboratory
Virus Concentration
Multiple concentration methods have been employed globally, with the most common being [47]:
- Polyethylene Glycol (PEG) Precipitation (24 studies)
- Centrifugation-based Methods (22 studies)
- Ultrafiltration (13 studies)
- Electronegative Membrane Filtration (4 studies)
- Aluminum-based Adsorption Precipitation (3 studies)
- Organic Flocculation (2 studies)
RNA Extraction
- Sample Type: Use concentrated wastewater solids or liquid samples
- Inhibitor Removal: Include enzymatic or chemical inhibition removal steps
- Extraction Method: Employ commercial nucleic acid extraction kits with appropriate controls
- Quality Assessment: Measure RNA concentration and purity; include process controls
Nested PCR Assay for SARS-CoV-2 Detection
The following protocol adapts the one-tube nested qPCR approach for SARS-CoV-2 detection in wastewater, based on methodologies successfully applied to brucellosis detection [49] and SARS-CoV-2 variant monitoring [51].
Primer and Probe Design
- Target Selection: Focus on conserved regions of SARS-CoV-2 genome (N, S, ORF1a genes)
- Outer Primers: Design to amplify 200-300 bp fragment
- Inner Primers: Design to amplify 100-150 bp fragment internal to first amplicon
- Probe Design: Develop hydrolysis probes (TaqMan) with distinct fluorophore-quencher pairs
- Specificity Validation: Verify using BLAST analysis against human and microbial genomes
One-Tube Nested qPCR Reaction Setup
- Reaction Volume: 20-25 μL total volume
- Reaction Components:
- 5-10 μL extracted RNA template
- 0.5 μM outer forward and reverse primers
- 0.3 μM inner forward and reverse primers
- 0.2 μM each probe
- 1× reaction buffer
- 3.5 mM MgCl₂
- 0.4 mM dNTPs
- 1.25 U DNA polymerase
- 0.5 U reverse transcriptase (for RT-PCR)
- Thermal Cycling Conditions:
- Reverse Transcription: 50°C for 15 minutes (if required)
- Initial Denaturation: 95°C for 3 minutes
- First Stage Amplification (10 cycles):
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 45 seconds
- Second Stage Amplification (35 cycles):
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 55°C for 45 seconds (with fluorescence detection)
Controls and Validation
- Positive Control: Synthetic SARS-CoV-2 RNA fragment
- Negative Control: Nuclease-free water
- Inhibition Control: Process control virus (e.g., Bovine Coronavirus Vaccine)
- Extraction Control: Pepper Mild Mottle Virus (PMMoV) as fecal indicator
- Calibration Curve: Serial dilutions of target RNA for quantification
Data Analysis and Interpretation
- Quantification: Use standard curves for absolute quantification or comparative Cq method for relative quantification
- Variant Identification: Analyze mutation patterns based on probe detection or sequencing results
- Normalization: Normalize target concentration to flow rate, population, or fecal indicators
- Trend Analysis: Apply statistical models to identify significant changes in viral concentrations
Research Reagent Solutions
Successful implementation of nested PCR for SARS-CoV-2 wastewater surveillance requires specific reagents and materials. The following table details essential research reagent solutions and their functions in the experimental workflow.
Table 3: Essential Research Reagents for Nested PCR-Based Wastewater Surveillance
| Reagent Category | Specific Examples | Function | Technical Considerations |
|---|---|---|---|
| Virus Concentration Reagents | Polyethylene glycol (PEG), Aluminum-based flocculants | Concentrate viral particles from large wastewater volumes | PEG precipitation offers cost-effectiveness; membrane filtration provides rapid processing |
| Nucleic Acid Extraction Kits | Commercial silica-based kits, Magnetic bead systems | Isolate viral RNA from complex wastewater matrices | Must include inhibitor removal steps; validated for wastewater samples |
| PCR Enzymes & Master Mixes | Reverse transcriptase, Thermostable DNA polymerase, dNTPs | Catalyze cDNA synthesis and DNA amplification | Select enzymes with high processivity and fidelity; optimize buffer composition |
| Primers & Probes | Outer primer set (200-300 bp), Inner primer set (100-150 bp), Dual-labeled hydrolysis probes | Target-specific amplification and detection | Design for variant discrimination; validate specificity and sensitivity |
| Inhibition Controls | Bovine Coronavirus Vaccine (BCoV), Pepper Mild Mottle Virus (PMMoV) | Monitor PCR inhibition and extraction efficiency | Should be added before nucleic acid extraction; use at consistent concentration |
| Quantification Standards | Synthetic SARS-CoV-2 RNA transcripts, G-block fragments | Generate standard curves for absolute quantification | Should encompass entire target sequence; verify concentration by multiple methods |
Discussion
Advantages of Nested PCR in WBE
The integration of nested PCR methodologies into wastewater surveillance programs addresses several critical challenges in environmental pathogen detection. The enhanced sensitivity of nested PCR, demonstrated by a 100-fold improvement over conventional qPCR in some applications [49], is particularly valuable for early outbreak detection when community prevalence remains low. This sensitivity enables public health officials to implement targeted interventions before widespread clinical transmission occurs.
The variant tracking capability of nested PCR approaches, especially when coupled with sequencing, provides crucial information about circulating SARS-CoV-2 lineages. The demonstration that wastewater sequencing could identify Omicron-associated mutations before official designation as a Variant of Concern [51] highlights the predictive potential of these methods. This advanced warning system allows healthcare systems to prepare for potential shifts in transmission dynamics, disease severity, or vaccine escape.
Implementation Considerations
While nested PCR offers significant advantages, successful implementation in public health laboratories requires addressing several practical considerations:
- Contamination Control: The high sensitivity of nested PCR increases vulnerability to amplicon contamination. The one-tube nested approach significantly mitigates this risk [49]
- Standardization: Inter-laboratory comparison requires standardized protocols, controls, and data normalization methods [46]
- Cost-Benefit Analysis: Although more complex than standard qPCR, the early warning provided by nested PCR may justify additional costs through averted outbreaks
- Data Integration: Wastewater data should be interpreted alongside clinical surveillance to provide comprehensive situational awareness [52]
Future Directions
The expanding application of WBE to other pathogens, including influenza, RSV, measles, and Candida auris [46], suggests a growing role for highly sensitive detection methods like nested PCR in public health practice. Future developments will likely focus on:
- Multiplexed Nested PCR Panels: Simultaneous detection of multiple pathogens in wastewater
- Automation: Development of integrated systems for sample processing, analysis, and data reporting
- Modeling Integration: Advanced statistical and machine learning approaches to translate wastewater signals into public health actionable information [52]
- Global Surveillance Networks: Expanded wastewater monitoring in resource-limited settings to enhance pandemic preparedness
Wastewater-based epidemiology represents a transformative approach to public health surveillance, offering unbiased, cost-effective monitoring of community disease transmission. The integration of nested PCR methodologies significantly enhances the sensitivity and specificity of SARS-CoV-2 detection in complex wastewater matrices, enabling more reliable variant tracking and earlier outbreak detection. As demonstrated by global monitoring efforts, this approach provides valuable data complementary to clinical surveillance systems, with particular utility for capturing signals from asymptomatic individuals and detecting emerging variants before clinical case recognition.
The continued refinement of nested PCR protocols, including the development of one-tube systems that reduce contamination risk, will further strengthen the role of WBE in public health practice. As the field advances, the integration of wastewater data with sophisticated modeling approaches [52] will enhance our ability to predict disease dynamics and implement targeted interventions. The ongoing expansion of WBE to other pathogens of public health concern suggests that nested PCR methodologies will play an increasingly important role in global disease surveillance infrastructure, contributing to more resilient public health systems capable of responding rapidly to emerging threats.
Optimizing Nested PCR Assays: Overcoming Inhibition and Variant Escape
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has highlighted a critical challenge in molecular diagnostics: the virus's persistent evolution and genetic variability can compromise the accuracy of detection assays. Within this context, multi-target assay combinations have emerged as a powerful strategy to enhance detection sensitivity and ensure diagnostic reliability. This approach is particularly valuable when integrated with established but technically demanding methods like nested PCR, which, despite its high sensitivity, faces challenges in standardization and throughput. This application note details how combining multi-target designs with sophisticated PCR methodologies creates a robust framework for SARS-CoV-2 detection, resilient against viral mutations and capable of supporting large-scale screening efforts.
Performance Comparison of SARS-CoV-2 Detection Assays
The diagnostic performance of an assay is fundamentally linked to its design, particularly the number of viral targets it detects. The following table summarizes the performance of various commercial and in-house assays as reported in recent studies.
Table 1: Comparative Performance of Selected SARS-CoV-2 Molecular Assays
| Assay Name / Type | Gene Targets | Sensitivity (%) | Specificity (%) | Key Characteristics | Source |
|---|---|---|---|---|---|
| Semi-nested Heptaplex RT-PCR | 7-plex melting analysis | 100 | 99.87 | LOD: 7.2 copies/reaction; enables 96-sample pooled testing | [53] |
| Tata MD CHECK RT-PCR XF | ORF1ab, N | 93.9 | 100 | Direct RT-PCR (no RNA extraction); rapid turnaround (~1 hour) | [19] |
| In-house SYBR-Green Method | Not specified | 91.2 | 90.9 | Consistent detection across Variants of Concern (VOCs) | [54] |
| Da An Gene Kit | N, ORF1ab | 68.4 | 100 | Performance varies significantly with viral variant | [54] |
| Sansure Kit | N, ORF1ab | 91.2 | 54.5 | High false-positive rate observed | [54] |
| TaqPath Kit | S, N, ORF1ab | 70.2 | 100 | 3-gene target; exhibits S-gene target failure (SGTF) in some VOCs | [54] |
The data demonstrates that assays incorporating multiple genetic targets generally achieve higher and more consistent sensitivity. For instance, the semi-nested heptaplex assay and the 3-gene TaqPath kit show robust performance, whereas kits with fewer targets are more susceptible to performance degradation from viral mutations [54]. The U.S. FDA notes that tests with multiple targets are "more likely to continue to perform well when new variants emerge" compared to single-target tests [55].
Table 2: Impact of Viral Load on Antigen Test Sensitivity
| Antibody Titre (BAU/mL) | Sensitivity with Single Antigen (%) | Sensitivity with Dual Antigen (NP & RBD) (%) |
|---|---|---|
| ≤ 25 | 86.0 | >96.0 |
| ≤ 7.5 | 57.0 | >96.0 |
| Source | [56] | [56] |
This principle extends to serological assays. A study on a dual-target lateral flow immunoassay showed that detecting IgG antibodies against both the nucleoprotein (NP) and receptor-binding domain (RBD) significantly enhanced sensitivity, especially in individuals with low antibody titers, achieving a combined diagnostic sensitivity of 96.1% [56].
Experimental Protocols
Protocol 1: Semi-Nested, Heptaplex RT-PCR with Melting Analysis for High-Throughput Detection
This protocol describes a highly sensitive and specific method for detecting SARS-CoV-2 RNA, suitable for individual or pooled sample testing [53].
Workflow
Key Steps
- Sample Collection and RNA Extraction: Collect nasopharyngeal swabs and place them in viral transport medium. Extract RNA using a validated magnetic bead-based or column-based nucleic acid extraction kit [53] [57].
- Semi-Nested Heptaplex Pre-amplification:
- First Round (RT-PCR): Perform a one-tube reverse transcription and multiplex PCR amplification using a primer set designed for seven different conserved regions of the SARS-CoV-2 genome.
- Second Round (qPCR): Use the product from the first round as a template for a quantitative real-time PCR. This step employs fluorescent probes or DNA-binding dyes to generate target-specific melting curves [53].
- Melting Curve Analysis and AI Interpretation: After amplification, run a high-resolution melting curve analysis. The complex melting spectrum generated from the heptaplex amplicons is interpreted using a dedicated artificial intelligence algorithm to confirm the presence of SARS-CoV-2 RNA and potentially identify specific variants [53].
- Pooled Testing Application: For high-throughput screening, this assay supports a 96-sample pooling strategy. Combine 5-10 µL from each of 96 extracted RNA samples into a single pool. Subject the pool to the semi-nested RT-PCR protocol. If the pool tests positive, individual samples can be re-tested from the pre-amplified product, allowing a single 96-well qPCR plate to screen up to 8,820 individual samples [53].
Protocol 2: Development of Variant-Specific RT-PCR Assays (e.g., for BA.2.86)
This protocol is for designing and validating targeted RT-PCR assays for specific SARS-CoV-2 variants, providing a rapid and cost-effective alternative to full genomic sequencing [58].
Workflow
Key Steps
- In Silico Design and Validation:
- Identify unique mutation profiles of the target variant (e.g., the >30 Spike protein mutations in BA.2.86) from genomic databases.
- Design primer and probe sets that specifically bind to regions encompassing these variant-defining mutations. Use tools like Primer-BLAST to ensure specificity.
- Perform in silico validation against a wide range of SARS-CoV-2 sequences to confirm that the assay will only detect the intended variant and its sublineages [58].
- Wet-Lab Validation with Control Materials:
- Obtain synthetic RNA or clinical samples confirmed by sequencing to contain the target variant (e.g., BA.2.86).
- Test the designed assays (e.g., PiroMet-1 and PiroMet-2 for BA.2.86) using standard RT-qPCR or digital RT-PCR conditions to confirm they yield a positive signal only with the correct variant [58].
- Determination of Analytical Sensitivity and Specificity:
- Perform limit of detection (LOD) studies using serial dilutions of quantified target RNA. The LOD for well-designed assays can be as low as 1-2 RNA copies/µL [58].
- Test against RNA from other common variants and respiratory pathogens to confirm analytical specificity and rule up cross-reactivity.
- Application in Surveillance:
- Apply the validated assay to clinical nasopharyngeal samples or RNA extracted from wastewater.
- For comprehensive surveillance, use the variant-specific assay in combination with a general SARS-CoV-2 assay (e.g., JRC-CoV-UCE.2) and an assay that detects other variants (e.g., OmMet for non-BA.2.86 Omicron). This multi-assay combination allows for the relative quantification and prevalence monitoring of the specific variant within a community [58].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Advanced SARS-CoV-2 PCR Assays
| Reagent / Kit | Function | Example Use Case |
|---|---|---|
| Multiplex RT-PCR Master Mix | Provides enzymes and optimized buffers for simultaneous amplification of multiple targets in a single reaction. | Essential for heptaplex semi-nested RT-PCR and other multi-target assays [53]. |
| Viral Nucleic Acid Extraction Kit | Isolves and purifies viral RNA from clinical samples (e.g., nasopharyngeal swabs, saliva). | Magnetic bead-based kits (e.g., Seegene STARMag) are widely used for high-throughput workflows [57]. |
| SARS-CoV-2 Positive Control RNA | Validates assay performance; used for determining the limit of detection (LOD) and standard curves. | Crucial for validating in-house and commercial assays, including variant-specific tests [58]. |
| DNA Intercalating Dye (e.g., SYBR Green) | Binds double-stranded DNA, enabling real-time quantification and melting curve analysis. | Used in the in-house SYBR-Green method that showed 91.2% sensitivity [54]. |
| Variant-Specific Primers & Probes | Oligonucleotides designed to uniquely identify mutations specific to a SARS-CoV-2 variant. | Core component of the PiroMet assays for BA.2.86 detection [58]. |
| Pooling Matrix / Diluent | A solution used to combine individual samples into a pool without inhibiting the PCR reaction. | Required for high-throughput pooled testing strategies like P-BEST and 96-sample pooling [57] [53]. |
Addressing PCR Inhibition in Complex Matrices like Wastewater
Polymerase chain reaction (PCR) inhibition presents a significant challenge in molecular detection, particularly when analyzing complex matrices such as wastewater. These samples contain a diverse array of organic and inorganic substances—including humic acids, metals, polysaccharides, lipids, and proteins—that can interfere with nucleic acid extraction, inhibit polymerase activity, and ultimately lead to false-negative results or an underestimation of target concentrations [59] [60] [61]. The reliability of wastewater-based epidemiology (WBE), which gained prominence during the COVID-19 pandemic for monitoring community-level transmission of SARS-CoV-2, is fundamentally dependent on effectively overcoming this inhibition [59] [62] [61]. This application note outlines proven strategies and detailed protocols to mitigate PCR inhibition, with a specific focus on enhancing the performance of nested PCR assays for SARS-CoV-2 detection in wastewater.
Understanding PCR Inhibition in Wastewater
The complex wastewater matrix introduces numerous substances that inhibit molecular assays through various mechanisms. Humic acids can mimic DNA and interfere with the polymerization process, while metal ions (e.g., calcium) compete with essential magnesium co-factors for DNA polymerase binding sites [59] [60]. Complex polysaccharides and proteins may co-precipitate with nucleic acids or sequester target templates, reducing amplification efficiency [59]. These inhibitors often persist through nucleic acid extraction protocols, necessitating specific post-extraction countermeasures for accurate detection and quantification [60] [61].
The impact of these inhibitors is particularly critical when using highly sensitive techniques like nested PCR, which involves two successive rounds of amplification to detect low-abundance targets such as SARS-CoV-2 RNA in diluted wastewater samples [24] [17]. While nested PCR offers superior sensitivity for variant surveillance [24], its multi-step nature makes it vulnerable to inhibitor carry-over, potentially compromising even this robust detection method.
Strategies for Overcoming PCR Inhibition
Chemical and Biological Enhancers
Adding specific enhancers directly to PCR reactions represents a straightforward approach to mitigating inhibition. These compounds function by binding inhibitors or stabilizing reaction components.
Table 1: PCR Enhancers and Their Applications
| Enhancer | Recommended Concentration | Mechanism of Action | Effectiveness |
|---|---|---|---|
| T4 gene 32 protein (gp32) | 0.2 μg/μL | Binds to single-stranded DNA, preventing inhibitor binding and stabilizing nucleic acids [59] | Most effective; eliminated false negatives in wastewater [59] |
| Bovine Serum Albumin (BSA) | 0.1-0.5 μg/μL | Binds to inhibitors like humic acids, reducing their interaction with polymerase [59] [60] | Significant improvement; removed inhibition in all tested samples [59] |
| Dimethyl Sulfoxide (DMSO) | 1-5% | Lowers DNA melting temperature, destabilizes secondary structures [59] | Variable effectiveness; concentration-dependent [59] |
| Formamide | 1-5% | Destabilizes DNA helix, facilitating primer binding [59] | Variable effectiveness; concentration-dependent [59] |
| TWEEN-20 | 0.1-1% | Non-ionic detergent that counteracts inhibitory effects on Taq polymerase [59] | Moderate improvement [59] |
| Glycerol | 1-5% | Protects enzymes from degradation and denaturation [59] | Moderate improvement [59] |
Physical and Chemical Removal Methods
Alternative approaches focus on physically removing inhibitors from samples prior to PCR amplification.
- Sample Dilution: A 10-fold dilution of extracted nucleic acids effectively reduces inhibitor concentration and eliminated false negative results in wastewater samples [59]. However, this approach simultaneously dilutes the target template, potentially reducing sensitivity for low-abundance targets [59] [60].
- Polymeric Adsorbents: Treatment with Supelite DAX-8 (5% w/v) effectively removes humic acids by permanently binding these compounds. This treatment has significantly improved viral detection in environmental waters without substantial viral loss [60].
- Commercial Inhibitor Removal Kits: These specialized kits utilize column matrices designed to remove polyphenolic compounds, humic acids, tannins, and other common PCR inhibitors. They have successfully removed inhibition in wastewater samples, though their effectiveness varies [59] [60].
Experimental Protocols for Inhibition Management
T4 Gene 32 Protein Enhancement Protocol
Principle: T4 gp32 binds to single-stranded nucleic acids, protecting templates and making them more accessible to polymerase while reducing inhibitor binding [59].
Procedure:
- Master Mix Preparation: Prepare a standard PCR master mix according to your nested PCR protocol [24] [17].
- Enhancer Addition: Add T4 gp32 to a final concentration of 0.2 μg/μL directly to the master mix [59].
- Reaction Setup: Dispense 23 μL of the enhanced master mix into each reaction tube.
- Template Addition: Add 2 μL of extracted wastewater nucleic acids.
- Amplification: Perform nested PCR amplification using previously established cycling conditions for SARS-CoV-2 detection [24].
DAX-8 Treatment Protocol for Inhibitor Removal
Principle: DAX-8 resin selectively binds humic acids and other phenolic inhibitors through hydrophobic interactions, physically removing them from solution [60].
Procedure:
- Resin Preparation: Obtain Supelite DAX-8 resin (Sigma-Aldrich).
- Sample Treatment: Add DAX-8 to concentrated wastewater samples to achieve 5% (w/v) concentration [60].
- Incubation: Mix samples continuously for 15 minutes at room temperature.
- Separation: Centrifuge at 8,000 rpm for 5 minutes at 4°C to pellet the insoluble resin with bound inhibitors.
- Supernatant Collection: Carefully transfer the clarified supernatant to a fresh tube for nucleic acid extraction.
- Viral Recovery Assessment: Include process controls to monitor potential viral loss during treatment.
Dilution Approach for Inhibition Control
Principle: Simple dilution reduces inhibitor concentrations below their effective threshold while potentially retaining detectable target levels, especially when using sensitive nested PCR methods [59].
Procedure:
- Baseline Detection: First attempt detection with undiluted nucleic acid extracts.
- Dilution Series Preparation: If inhibition is suspected, prepare a dilution series (1:2, 1:5, 1:10) of extracted nucleic acids in nuclease-free water.
- Parallel Amplification: Amplify each dilution alongside the original extract using your nested PCR assay.
- Result Interpretation: Compare amplification efficiency across dilutions. Typically, a 10-fold dilution sufficiently reduces inhibition while maintaining target detectability in wastewater matrices [59].
The Scientist's Toolkit
Table 2: Essential Research Reagents for Inhibition Management
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| PCR Enhancers | T4 gene 32 protein (gp32), Bovine Serum Albumin (BSA) | Binds inhibitors in the reaction mix; added directly to PCR [59] |
| Sample Diluents | Nuclease-free water, TE buffer | Dilutes inhibitor concentration in nucleic acid extracts [59] |
| Polymeric Adsorbents | Supelite DAX-8, Polyvinylpyrrolidone (PVP) | Removes humic acids and phenolic compounds prior to extraction [60] |
| Inhibitor-Resistant Kits | Commercial inhibitor removal kits | Column-based removal of PCR inhibitors from nucleic acids [59] [60] |
| Inhibition Controls | Murine norovirus (MNV), process controls | Monitors inhibition extent and method effectiveness [60] |
Effective management of PCR inhibition is fundamental to obtaining reliable results from wastewater-based surveillance programs. For nested PCR detection of SARS-CoV-2, incorporating systematic inhibition control measures—particularly the addition of T4 gp32 or BSA to reactions, implementing appropriate sample dilution strategies, or utilizing adsorbents like DAX-8—significantly enhances detection sensitivity and accuracy [24] [59] [60]. These protocols provide researchers with practical approaches to overcome the challenges posed by complex matrices, ensuring robust molecular detection in wastewater and other inhibitor-rich environmental samples.
The accuracy of reverse transcription polymerase chain reaction (RT-PCR), the gold standard diagnostic test for SARS-CoV-2, is fundamentally dependent on the precise complementarity between oligonucleotide components (primers and probes) and their target genomic sequences [63]. As an RNA virus, SARS-CoV-2 undergoes continuous evolution, characterized by the emergence of Variants of Concern (VOCs) with numerous mutations across their genome [64] [65]. These mutations can occur in the primer and probe binding regions, leading to mismatches that reduce hybridization efficiency and potentially cause false-negative results or decreased test sensitivity [64] [63] [66]. This application note details the impact of such mismatches on diagnostic efficacy and outlines validated protocols for the ongoing monitoring and adaptation of molecular assays within the critical context of nested PCR assay research for SARS-CoV-2 detection.
The Impact of Genetic Variation on PCR Assay Performance
Genetic variations in SARS-CoV-2, particularly within VOCs, directly challenge PCR assay performance by introducing mismatches in primer/probe binding sites. The following table summarizes the extensive mismatch analysis performed on publicly available PCR assays.
Table 1: Mismatch analysis in SARS-CoV-2 PCR assay target regions
| Target Gene | Number of Primers/Probes Analyzed | Group A (All except Omicron) Sequences with ≥1 Mismatch | Group B (Omicron only) Sequences with ≥1 Mismatch | Examples of Affected Assays |
|---|---|---|---|---|
| Nucleocapsid (N) | 36 | 87.7% (64/73 total) | 32.9% (24/73 total) | CDC (N1, N2), Charité Hospital [64] |
| Spike (S) | 22 | 87.7% (64/73 total) | 32.9% (24/73 total) | Various S-gene targeting assays [64] [66] |
| RdRp/Helicase | 15 | 87.7% (64/73 total) | 32.9% (24/73 total) | Corman-RdRp, Won-RdRp-1 [64] [63] |
The consequences of these mismatches are not merely theoretical. Specific examples include:
- N1 Probe Dropout: Mutations in the N1 probe binding region of the CDC qPCR assay led to a significant underestimation of viral load in wastewater surveillance, with a single mutation causing up to a 2.3-fold drop in signal [67].
- S-Gene Target Failure (SGTF): The accumulation of mutations in the Spike gene of the Alpha and Omicron VOCs caused a characteristic "S-gene dropout" in certain multiplex assays, serving as an early indicator of these variants but also highlighting a vulnerability in detection [66].
- Assay Performance Deterioration: An in-silico evaluation of 73 commercial qRT-PCR kits used in India found several kits to be unsatisfactory for detecting Delta and Omicron VOCs due to primer-probe mismatches [66].
Experimental Protocols for Mismatch Evaluation and Variant Detection
In-silico Screening of Primer and Probe Performance
A robust two-step in-silico screening process allows for the preemptive evaluation of PCR assay performance against current and future VOCs [66].
Table 2: Two-step in-silico screening protocol for primer/probe evaluation
| Step | Description | Key Tools & Databases | Critical Screening Criteria |
|---|---|---|---|
| 1. Data Curation | Retrieve and cluster SARS-CoV-2 VOC genome sequences to obtain a non-redundant dataset. | GISAID, CD-HIT software [63] [66] | Use complete genomes with <1% ambiguous bases (Ns). |
| 2. Sequence Alignment | Perform local alignment of primer/probe sequences against the curated genome database. | BLASTn, EMBOSS Water (Smith-Waterman algorithm) [66] | Identify percentage alignment, mismatches, and gaps. |
| 3. Mismatch Analysis | Parse results to determine position-specific mismatches across the length of the oligonucleotides. | Custom scripts, EMBOSS Water output [66] | Flag mismatches at the 3' end, contiguous mismatches, and central probe mismatches. |
Screening Criteria: A primer or probe is deemed acceptable only if it meets all of the following criteria [66]:
- Alignment Identity: ≥95% alignment with the VOC genome sequences.
- Full-length Match: The entire primer/probe length must match in ≥95% of aligned sequences.
- 3'/5' End Integrity: No mismatches at the 3' or 5' terminal nucleotides.
- Contiguous Matches: No more than three contiguous nucleotide mismatches.
- Probe Central Region: No single nucleotide mismatch in the central region of the probe (positions -1, 0, +1 for a ~20nt probe).
Nested PCR and Sanger Sequencing for Variant Identification
For wet-lab confirmation and specific variant identification, a nested PCR protocol followed by Sanger sequencing provides a cost-effective and reliable method.
Protocol: Nested PCR and Sequencing for VOC Identification [24]
Sample Preparation:
- Input: Throat swab or sputum samples from confirmed COVID-19 patients.
- RNA Extraction: Use an automated nucleic acid extraction system (e.g., EX3600, Zhijiang Ltd.) with 300 μL of sample input, eluting in 50 μL RNase-free water.
- Reverse Transcription: Convert 10 μL of extracted RNA to cDNA using a reverse transcriptase PCR kit (e.g., Mighty Script Plus Mix, Sangon Biotech) [24].
Primer Design:
- Target Regions: Focus on genomic regions with high mutation density relevant to circulating VOCs, such as the Spike (S), Nucleocapsid (N), and ORF1a genes [24] [68].
- Bioinformatic Tools: Utilize databases (GISAID, CNCB) and software (Oligo 7, NCBI Primer-BLAST) to design nested primer sets targeting specific mutations (e.g., K1973R in ORF1a, F59S in Spike) [24].
Nested PCR Amplification:
- First PCR:
- Reaction Mix: 2× Reaction Mix (25 μL), Taq Mix (2 μL), forward and reverse outer primers (1 μL each, 10 μM), RNase-free water (16 μL), cDNA template (5 μL).
- Cycling Conditions: 45°C for 30 min; 94°C for 2 min; 40 cycles of (94°C for 15 s, 55°C for 30 s, 68°C for 15 s); 68°C for 5 min.
- Second (Nested) PCR:
- Template: Diluted product from the first PCR (e.g., 1:1000 dilution).
- Reaction Mix: Similar to the first PCR, but using inner primers.
- Cycling Conditions: Optimize annealing temperature based on inner primer Tm (typically 48-50°C) [24].
Variant Identification:
- Sanger Sequencing: Purify the nested PCR products and subject them to Sanger sequencing.
- Analysis: Analyze the resulting sequences by aligning them to a reference genome (e.g., MN908947) to identify specific mutations and assign the lineage [24].
Diagram 1: Nested PCR workflow for VOC identification.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential reagents and tools for adapting PCR assays to VOCs
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| Automated Nucleic Acid Extractor | Standardizes RNA extraction from diverse sample types (swabs, wastewater). | Ensures high-quality template for downstream PCR [24] [69]. |
| One-Step RT-PCR Kits | Integrates reverse transcription and PCR amplification in a single reaction. | Streamlines the first amplification step in nested PCR protocols [69]. |
| Ultra-Conserved Element (UCE) Assays | Targets genomic regions with exceptionally low mutation rates. | Provides a robust "universal" detection method for current and future VOCs (e.g., JRC-CoV-UCE.1/2 assays) [65]. |
| Sanger Sequencing Reagents | Enables definitive identification of nucleotide sequences. | Confirms the presence of specific mutations in PCR amplicons for lineage assignment [24]. |
| Bioinformatics Tools (e.g., varVAMP) | Designs degenerate primers for highly variable viruses, minimizing mismatches. | Creates pan-specific primer schemes for sequencing or qPCR of diverse pathogens like SARS-CoV-2 [70]. |
The relentless genetic evolution of SARS-CoV-2 necessitates a paradigm of continuous vigilance and adaptation in molecular diagnostics. The systematic evaluation of primer and probe mismatches, facilitated by the in-silico and wet-lab protocols detailed herein, is paramount for maintaining diagnostic accuracy. Integrating these strategies, particularly within nested PCR frameworks and leveraging ultra-conserved genomic targets, empowers researchers and public health officials to rapidly respond to the ongoing challenge of viral evolution, ensuring reliable detection and surveillance of SARS-CoV-2 now and in the future.
Analyzing Cycle Threshold (Ct) Values for Qualitative and Quantitative Assessment
In quantitative PCR (qPCR) and reverse transcription quantitative PCR (RT-qPCR), the cycle threshold (Ct) value represents the number of nucleic acid amplification cycles required for the signal generated by target amplification to cross a predetermined threshold [71]. This value is inversely related to the amount of target nucleic acid in the sample; lower Ct values indicate higher viral loads, while higher Ct values correspond to lower viral loads [71]. In the context of SARS-CoV-2 detection, Ct values have served as a semi-quantitative measure of viral load, providing valuable information for both clinical management and public health surveillance [72] [4].
The interpretation of Ct values requires careful consideration of multiple technical and biological factors. Results can be influenced by pre-analytical variables such as swabbing technique, sample storage conditions, and RNA extraction methods, as well as analytical factors including PCR reagents, instrumentation, and the specific gene targets being amplified [71] [73]. Despite these limitations, when measured with a standardized assay, population-level Ct values can provide crucial information about epidemic growth and variant emergence [72].
The Role of Ct Values in SARS-CoV-2 Research
Correlation with Viral Load and Clinical Parameters
Ct values have demonstrated significant correlations with various clinical and epidemiological parameters. During the COVID-19 pandemic, lower Ct values (indicating higher viral loads) were associated with testing just after symptom onset, presence of classic COVID-19 symptoms, increased duration of viral shedding, and higher risk of severe illness [72]. Additionally, Ct values have served as a useful proxy for infectiousness, with lower values indicating higher transmission potential [72] [74].
Population-level analysis of Ct values has proven valuable for public health surveillance. A study analyzing daily mean Ct values in England found that spike (S) gene target-specific mean Ct values decreased 6–29 days before positive test counts increased, providing an early indication of emerging variants such as Delta and Omicron [72]. This approach enabled real-time monitoring of variant-driven epidemic waves based on Ct value trends.
Ct Value Interpretation Challenges
Despite their utility, Ct values present significant interpretation challenges. Cq values are not a definitive measure as they depend on subjective data analysis and should not be used for rigid cut-off settings [73]. A Ct value on its own should never be used to compare results obtained with different instruments, reagents, or at different times [73]. This limitation has led many diagnostic tests to provide qualitative (present/absent) results rather than quantitative Ct values [73].
The relationship between Ct values and viral load is further complicated by mutations in primer and probe binding sites. Discrepancies in Ct values between different gene targets (e.g., N2 and E genes) can indicate emerging variants with characteristic mutations, as demonstrated during a hospital cluster analysis where an approximately 10-cycle difference between targets revealed a specific point mutation (G29179T) [75].
Nested PCR Methodology for Enhanced SARS-CoV-2 Detection
Principles of Nested PCR
Nested PCR is a technique that significantly enhances detection sensitivity through two successive amplification rounds using two sets of primers [4]. The initial round amplifies a larger target region, while the second round uses "nested" primers that bind internally to the first amplicon, resulting in exponential increase in specificity and sensitivity [1]. This method is particularly valuable for detecting low viral loads where conventional RT-PCR may yield false negatives or require lower Ct value thresholds [10].
For SARS-CoV-2 detection, the nucleocapsid (N) gene serves as an ideal target due to its high conservation, stability, and fewer mutations compared to other genomic regions [4]. The enhanced sensitivity of nested PCR makes it particularly suitable for surveillance in animal populations and human samples with low viral loads, providing a cost-effective alternative to real-time RT-PCR [10] [4].
Detailed Nested PCR Protocol for SARS-CoV-2 Detection
Primer Design and Optimization
Primer Sequences Targeting the N Gene:
- External Forward Primer (Ext2019nCorVF): 5'-GGCAGTAACCAGAATGGAGA-3' (position 28346-28365)
- External Reverse Primer (Ext2019nCorVR): 5'-CTCAGTTGCAACCCATATGAT-3' (position 28681-28661)
- Internal Forward Primer (intF): 5'-CACCGCTCTCACTCAACAT-3' (position 28432-28450)
- Internal Reverse Primer (intR): 5'-CATAGGGAAGTCCAGCTTCT-3' (position 28643-28624) [1]
Primer Optimization:
- Optimal primer concentration: 5 pmol/μL for each primer [4]
- Annealing temperature for first round: 49°C [4]
- Annealing temperature for second round: 51°C [4]
- Alternative annealing temperature: 54.6°C for both rounds (as validated in another study) [1]
Sample Preparation and RNA Extraction
- Sample Collection: Collect oropharyngeal, nasopharyngeal, or conjunctival swabs and place in universal transport medium (UTM) or viral transport medium [1] [75].
- RNA Extraction: Use commercial RNA extraction kits such as the ISOLATE II RNA Mini kit following manufacturer's instructions [1]. Automated systems like QIAcube can also be employed for consistent results [75].
- RNA Quantification and Quality Control: Analyze RNA extracts using spectrophotometry (e.g., NanoDrop 2000) to ensure adequate concentration and purity [1].
Reverse Transcription and Amplification
cDNA Synthesis:
- Use 7μL of extracted RNA template
- Employ SensiFAST cDNA synthesis kit or equivalent
- Reaction conditions: 25°C for 10 min, 42°C for 15 min, 80°C for 5 min [1]
First Round PCR Amplification:
- Reaction volume: 25μL
- Composition: 12.5μL My Taq HS red mix, 4μL cDNA, 1μL each external primer (10 pmol/μL), 6.5μL PCR grade water
- Cycling conditions: Initial denaturation at 95°C for 1 min; 35 cycles of 95°C for 15s, 49-54.6°C for 15s, 72°C for 10s; final extension at 72°C for 5 min [1] [4]
Second Round PCR Amplification:
- Reaction volume: 25μL
- Composition: 12.5μL My Taq HS red mix, 0.5μL first PCR product, 1μL each internal primer (10 pmol/μL), 10μL PCR grade water
- Cycling conditions: Same as first round with appropriate annealing temperature [1]
Product Analysis and Validation
- Gel Electrophoresis: Run 5-10μL of second round PCR product on 2% agarose gel with ethidium bromide staining
- Visualization: Examine under UV transilluminator for expected band size (212bp with above primers) [1]
- Sequencing Validation: Perform Sanger sequencing using Big Dye Terminator kit and capillary sequencer for confirmatory analysis [1]
Comparative Performance Analysis: Nested PCR vs. Real-Time RT-PCR
Sensitivity and Specificity Assessment
Table 1: Performance Metrics of Nested PCR for SARS-CoV-2 Detection
| Parameter | Performance | Experimental Conditions |
|---|---|---|
| Sensitivity | 95% | Compared to real-time RT-PCR with animal samples [10] |
| Specificity | 100% | No cross-reactivity with CCV, FIPV, or other coronaviruses [10] [4] |
| Limit of Detection | ~50 copies/μL | Corresponding to Ct value of 31.5 in real-time RT-PCR [10] |
| Detection Range | Ct 17-31.5 | Equivalent to high, moderate, and low viral loads [10] |
| Inter-assay Agreement | Kappa = 0.829 | Excellent agreement with real-time RT-PCR [10] |
Ct Value Correlation with Viral Load Categories
Table 2: Ct Value Interpretation Guide for Viral Load Assessment
| Ct Value Range | Viral Load Category | Infectiousness Potential | Nested PCR Detection |
|---|---|---|---|
| < 24 | High | High | Consistently detected [4] |
| 25-29 | Moderate | Moderate | Consistently detected [4] |
| 30-35 | Low | Low | Detected (∼95% sensitivity) [10] [4] |
| > 35 | Very Low | Minimal | Variable detection [74] |
Recent research has demonstrated that nested PCR effectively detects SARS-CoV-2 in samples with Ct values as high as 31.5 (equivalent to approximately 50 copies/μL), making it particularly valuable for surveillance of low viral load infections [10]. The technique shows no cross-reactivity with other pathogenic coronaviruses such as canine coronavirus (CCV) and feline infectious peritonitis virus (FIPV), confirming its high specificity [4].
Research Reagent Solutions for SARS-CoV-2 Detection
Table 3: Essential Research Reagents for SARS-CoV-2 Nested PCR
| Reagent Category | Specific Products | Application Function |
|---|---|---|
| RNA Extraction Kits | ISOLATE II RNA Mini Kit | Viral RNA purification from swab samples [1] |
| cDNA Synthesis Kits | SensiFAST cDNA Synthesis Kit | Reverse transcription of RNA to DNA template [1] |
| PCR Master Mixes | My Taq HS Red Mix | High-sensitivity amplification with visible dye [1] |
| Positive Controls | SARS-CoV-2 USA/WA1/2020 | Assay validation and sensitivity determination [1] |
| Primer Sets | N gene-specific primers | Target amplification with high specificity [1] [4] |
| Electrophoresis Reagents | Agarose, Ethidium Bromide | PCR product visualization and size confirmation [1] |
| Sequencing Kits | BigDye Terminator v3.1 | Amplicon sequence verification [1] [75] |
Application in SARS-CoV-2 Surveillance and Research
Animal Model Surveillance
Nested PCR provides a cost-effective solution for large-scale animal surveillance studies, which are crucial for understanding the zoonotic potential of SARS-CoV-2. The technique has been successfully applied to detect natural infections in companion animals such as cats and dogs, with studies confirming infections during the first wave of human infections in multiple countries [1] [10]. The method's high sensitivity enables detection of low viral loads that might be missed by conventional PCR methods, providing crucial data for understanding cross-species transmission dynamics [4].
Variant Detection and Monitoring
The combination of Ct value analysis and targeted PCR methods has proven valuable for variant monitoring. Specific Ct value patterns, such as S-gene target failure (SGTF) observed in TaqPath assays, provided early indication of emerging variants including Alpha and Omicron [72]. Discrepancies in Ct values between different gene targets can signal mutations in primer binding regions, enabling preliminary variant identification without the need for full genome sequencing [75] [54].
Experimental Workflow Visualization
Nested PCR Workflow for SARS-CoV-2 Detection
Quality Control and Technical Considerations
Standard Curve Implementation
For quantitative applications, establishing a standard curve is essential:
- Reference Standards: Use accurately quantified control standards
- Linear Range: Plot Cq values against log-transformed gene copy numbers
- Efficiency Calculation: Apply formula E = -1 + 10^(-1/slope)
- Sensitivity Determination: Y-intercept provides theoretical detection limit [73]
Inhibition Controls and Validation
Include appropriate controls to detect amplification inhibitors:
- Internal extraction controls
- Positive and negative amplification controls
- Cross-reactivity panels against related pathogens [1] [4]
The integration of Ct value analysis with nested PCR methodologies provides a powerful approach for SARS-CoV-2 detection and quantification. This combination offers enhanced sensitivity for low viral load detection while maintaining cost-effectiveness for large-scale surveillance studies. The structured protocols and analytical frameworks presented in this application note provide researchers with validated methodologies to advance SARS-CoV-2 research, particularly in resource-limited settings and animal surveillance programs where cost-effective sensitive detection is paramount.
Benchmarking Performance: Nested PCR vs. ddPCR and Commercial Kits
In the context of SARS-CoV-2 detection, the analytical sensitivity of a molecular assay, typically defined by its Limit of Detection (LOD), is a critical performance parameter. It represents the lowest concentration of viral nucleic acid that can be reliably detected by the assay. The ongoing pandemic has highlighted the need for highly sensitive diagnostic methods, particularly for screening asymptomatic individuals or for detecting infections during the low viral load phases of disease. While quantitative real-time PCR (qRT-PCR) remains the gold standard, digital droplet PCR (ddPCR) and novel nested PCR strategies have emerged as powerful alternatives, each with distinct advantages in sensitivity and precision. This Application Note details the comparative analytical sensitivity of these methods, providing a framework for researchers and drug development professionals to select and optimize detection assays for SARS-CoV-2 and other pathogens.
Comparative LOD Performance of Detection Technologies
Direct comparisons of qRT-PCR, ddPCR, and nested PCR methods reveal significant differences in their analytical sensitivity for detecting SARS-CoV-2. The following table summarizes key performance metrics from recent studies.
Table 1: Comparative Analytical Sensitivity of SARS-CoV-2 Detection Methods
| Detection Method | Target Gene(s) | Reported LOD (copies/mL) | Key Comparative Finding | Reference |
|---|---|---|---|---|
| One-Step Nested (OSN) qRT-PCR | ORF1ab, N | 189.1 - 194.7 | Highest positive rate (82.35%) in clinical samples; superior to ddPCR and qRT-PCR | [30] |
| Droplet Digital PCR (ddPCR) | ORF1ab, N | 336.8 - 401.8 | More sensitive than qRT-PCR; positive rate of 67.65% in clinical samples | [30] |
| Quantitative RT-PCR (qRT-PCR) | ORF1ab, N | 528.1 - 520.1 | Baseline method with 58.82% positive rate in clinical samples | [30] |
| Semi-nested RT-PCR | Multi-target | 7.2 copies/reaction | High sensitivity and specificity (100% and 99.87%); enables high-throughput pooled screening | [53] |
| Commercial qRT-PCR Kits | Varies (e.g., ORF1ab, N, S) | 68 - 2264 | Performance varies significantly by manufacturer and target gene | [36] |
This comparative data demonstrates that nested PCR formats, particularly OSN-qRT-PCR, achieve the lowest LOD, making them particularly suitable for detecting low viral loads where standard qRT-PCR may yield false negatives [30]. The absolute quantification capabilities of ddPCR also make it a valuable tool for sensitive detection and for validating other methods [76].
Experimental Protocols for LOD Determination
To ensure reliable and comparable results, standardized protocols for determining the LOD of each method are essential. The following sections outline established experimental workflows.
Protocol for LOD Assessment Using Reference Materials
This protocol is adapted from studies comparing qRT-PCR and ddPCR for viral load quantification [77] [76].
1. Sample Preparation and Serial Dilution:
- Obtain quantified standard reference material, such as armored RNA, pseudoviral RNA, or viral culture supernatant [76] [30] [36].
- Perform serial log dilutions (e.g., 10-fold or 0.5-log) in a matrix that mimics the clinical sample, such as universal transport media (UTM) or nuclease-free water [76]. The dilution series should span the expected detection limit of the assays being evaluated.
- Aliquot and store dilutions at -80°C to minimize freeze-thaw cycles [76].
2. Nucleic Acid Extraction:
- Extract nucleic acids from a standardized volume (e.g., 200-400 µL) of each dilution using a commercial viral RNA/DNA kit [77] [76].
- Include an internal control during extraction to monitor efficiency and potential inhibition [77].
- Elute in a consistent, small volume (e.g., 50-90 µL) to maximize target concentration.
3. Parallel Amplification and Detection:
- Test each dilution level with a sufficient number of replicates (e.g., n=20) across the different platforms (qRT-PCR, ddPCR, nested PCR) [76].
- For qRT-PCR: Perform amplification following manufacturer protocols. Use a calibration curve for quantification [77].
- For ddPCR: Prepare reaction mix and generate droplets. After endpoint PCR, read the plate on a droplet reader and analyze using Poisson statistics for absolute quantification without a standard curve [77] [30].
- For Nested PCR: Perform the first amplification round, followed by a second round using inner primers. Detection can be via real-time fluorescence or melting curve analysis [53].
4. Data Analysis and LOD Calculation:
- The experimental LOD is defined as the lowest concentration at which ≥95% of replicates test positive [76].
- For greater statistical rigor, perform Probit analysis on the results from concentrations around the detection limit to determine the LOD with 95% confidence [76].
Protocol for Clinical Sample Verification
1. Sample Collection:
- Collect confirmed positive clinical samples (e.g., nasopharyngeal swabs) representing a range of viral loads, including low-positive samples [30] [54].
2. Head-to-Head Comparison:
- Extract nucleic acids from all samples and test them in parallel using the methods being compared (e.g., qRT-PCR, ddPCR, OSN-qRT-PCR) [30].
- Calculate and compare the positive detection rates for each method, particularly for samples with low viral loads [30].
Workflow and Logical Relationships
The decision to use a particular detection technology involves trade-offs between sensitivity, throughput, cost, and complexity. The following diagram illustrates the logical decision-making process for selecting an appropriate method based on research objectives.
Figure 1: A decision workflow for selecting a SARS-CoV-2 detection method based on research priorities, highlighting the role of nested PCR for achieving maximum sensitivity.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists essential reagents and their functions for implementing the PCR methodologies discussed in this note.
Table 2: Key Research Reagents for SARS-CoV-2 PCR Detection Assays
| Reagent / Kit | Function / Application | Example Use Case |
|---|---|---|
| International Standard Reference Materials (e.g., WHO International Standard) | Provides a universally accepted unitage (IU) for assay calibration and comparison | Harmonizing viral load measurements across different labs and platforms [77] |
| Pseudoviral RNA | Safe, non-infectious positive control containing target sequences of SARS-CoV-2 | Determining analytical sensitivity (LOD) and validating assay performance without BSL-3 requirements [30] |
| Viral Nucleic Acid Extraction Kits (e.g., Qiagen EZ1 virus kit) | Isolation and purification of viral RNA from clinical or environmental samples | Preparing template RNA for downstream qRT-PCR, ddPCR, or nested PCR amplification [77] [76] |
| One-Step RT-ddPCR Advanced Kit | Reverse transcription and droplet digital PCR in a single reaction mix | Absolute quantification of SARS-CoV-2 RNA copy number without a standard curve [78] |
| One-Step Nested RT-PCR Reagents | Enzymes and buffers supporting two sequential amplifications in a single tube | Highly sensitive detection of SARS-CoV-2 in samples with low viral load [30] |
| Restriction Enzymes (e.g., HaeIII, EcoRI) | Digests genomic DNA to improve access to target genes | Enhancing precision and accuracy in ddPCR, especially for targets with high copy numbers [79] |
Within the broader research on nested PCR assays for SARS-CoV-2 detection, the clinical validation of sensitivity and specificity is paramount. This application note consolidates robust experimental data from multiple studies to demonstrate the high diagnostic accuracy of nested PCR methodologies in both human and animal patient samples. The technique's exceptional performance, particularly in detecting low viral loads where conventional real-time RT-PCR may fail, establishes its utility for rigorous clinical and surveillance applications [1] [4] [80].
Data aggregated from independent clinical studies consistently report that nested PCR assays achieve high sensitivity and specificity for SARS-CoV-2 detection. The following table summarizes the key performance metrics from these validation studies.
Table 1: Clinical Performance of Nested PCR for SARS-CoV-2 Detection
| Study Description | Sensitivity | Specificity | Limit of Detection (LoD) | Sample Type |
|---|---|---|---|---|
| Validation against IVD kits (45 positive, 45 negative samples) [1] | 100% | 100% | 0.015 ng/μL RNA | Human and cat samples |
| Detection in animal samples (15 positive, 125 negative) [4] [10] | ~95% | 100% | ~50 copies/μL (Ct 31.5) | Dog and cat oropharyngeal swabs |
| Identification of false negatives (184 symptommatic, RT-PCR negative samples) [80] | 14.6% positive rate for ORF1ab (via nested PCR) | Not Applicable | Not Specified | Human nasopharyngeal swabs |
Detailed Experimental Protocol
This section details a standardized protocol for a two-round nested PCR targeting the SARS-CoV-2 N gene, validated for use with human and animal respiratory samples [1] [4].
Sample Processing and RNA Extraction
- Sample Collection: Collect nasopharyngeal or oropharyngeal swabs and place them in Viral Transport Medium (VTM).
- RNA Extraction: Perform nucleic acid extraction using a commercial silica-membrane based kit, such as the ISOLATE II RNA Mini Kit. Elute RNA in 60-100 μL of elution buffer.
- Quality Control: Quantify and assess the purity of the extracted RNA using a spectrophotometer (e.g., NanoDrop 2000). Store RNA at -80°C if not used immediately.
cDNA Synthesis
- Prepare Reaction Mix: Combine the following components in a nuclease-free tube:
- 7 μL of extracted RNA
- 8 μL of DEPC-treated water
- 4 μL of TransAmp buffer (or similar 5X RT buffer)
- 1 μL of reverse transcriptase enzyme (e.g., SensiFAST cDNA synthesis kit)
- Run Synthesis Program: Incubate the reaction mix in a thermal cycler using the following conditions:
- 25°C for 10 minutes (priming)
- 42°C for 15 minutes (reverse transcription)
- 80°C for 5 minutes (enzyme inactivation)
- Storage: Synthesized cDNA can be stored at -10°C to -20°C for short-term use.
Nested PCR Amplification
This protocol uses two sets of primers targeting the conserved N gene to enhance sensitivity and specificity [1]. The primer sequences are listed in Table 2.
Table 2: Primer Sequences for Nested PCR Targeting the N Gene
| Primer Name | Sequence (5' → 3') | Position (MN908947.3) | Amplicon Size |
|---|---|---|---|
| External Forward | GGCAGTAACCAGAATGGAGA | 28346-28365 | 335 bp |
| External Reverse | CTCAGTTGCAACCCATATGAT | 28681-28661 | |
| Internal Forward | CACCGCTCTCACTCAACAT | 28432-28450 | 212 bp |
| Internal Reverse | CATAGGGAAGTCCAGCTTCT | 28643-28624 |
First Round PCR (External Amplification)
- Prepare Reaction Mix (25 μL total volume):
- 12.5 μL of 2X My Taq HS Red Mix
- 1 μL of External Forward Primer (10 pmol/μL)
- 1 μL of External Reverse Primer (10 pmol/μL)
- 4 μL of cDNA template
- 6.5 μL of PCR-grade water
- Amplification Profile:
- Initial Denaturation: 95°C for 2 min
- 35 Cycles:
- Denaturation: 94°C for 30 sec
- Annealing: 49-54°C for 30 sec
- Extension: 72°C for 30 sec
- Final Extension: 72°C for 5 min
Second Round PCR (Internal Amplification)
- Prepare Reaction Mix (25 μL total volume):
- 12.5 μL of 2X My Taq HS Red Mix
- 1 μL of Internal Forward Primer (10 pmol/μL)
- 1 μL of Internal Reverse Primer (10 pmol/μL)
- 0.5 μL of first-round PCR product (use a diluted aliquot, e.g., 1:10 to 1:100, to minimize carryover of primers)
- 10 μL of PCR-grade water
- Amplification Profile: Use the same thermocycling conditions as the first round, though the annealing temperature may be optimized (e.g., 51°C).
Analysis of PCR Products
- Gel Electrophoresis: Load 5-10 μL of the second-round PCR product onto a 2% agarose gel stained with ethidium bromide. Include a appropriate DNA ladder.
- Visualization: Run the gel at 120-150 V for 30 minutes and visualize under a UV transilluminator. A positive result is indicated by a clear band of the expected size (212 bp).
Workflow Diagram
The following diagram illustrates the complete nested PCR experimental workflow.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials
| Reagent/Material | Function/Application | Example Product/Catalog |
|---|---|---|
| Viral RNA Extraction Kit | Isolation of high-quality viral RNA from swab samples | ISOLATE II RNA Mini Kit (Bioline) [1] |
| cDNA Synthesis Kit | Reverse transcription of viral RNA into stable cDNA | SensiFAST cDNA Synthesis Kit (Bioline) [1] |
| Hot-Start PCR Master Mix | High-fidelity DNA amplification with reduced non-specific background | My Taq HS Red Mix (Bioline) [1] |
| Nested Primers (N gene) | Specific amplification of SARS-CoV-2 nucleocapsid gene in two rounds | Custom oligonucleotides [1] [4] |
| Agarose Gel Electrophoresis System | Size-based separation and visualization of PCR amplicons | Standard laboratory setup with ethidium bromide stain [1] [80] |
Comparative Analysis of Commercial Kits and In-House SYBR-Green Methods
The unprecedented scale of the COVID-19 pandemic created a massive global demand for reliable, scalable, and affordable SARS-CoV-2 diagnostic testing. While TaqMan probe-based commercial real-time PCR kits emerged as the gold standard, their high cost and supply chain limitations prompted the development of economical alternatives, particularly in resource-limited settings [81] [82]. In-house SYBR Green-based methods present a viable, cost-effective option, though they require careful optimization and validation to ensure performance comparable to commercial kits [83] [84]. This application note provides a comparative analysis of these two approaches, focusing on their application in SARS-CoV-2 detection within the broader context of developing nested PCR assays. We present standardized protocols and performance metrics to guide researchers, scientists, and drug development professionals in selecting and implementing the most appropriate molecular diagnostic strategy for their specific needs.
Performance Comparison: Commercial Kits vs. In-House SYBR Green Methods
Extensive validation studies have demonstrated that well-optimized in-house SYBR Green assays can achieve performance metrics close to those of commercial TaqMan-based kits.
Table 1: Diagnostic Performance of SYBR Green vs. TaqMan Methods
| Evaluation Metric | Commercial TaqMan Kits | In-House SYBR Green Methods | Research Context |
|---|---|---|---|
| Sensitivity | Gold Standard Reference | 84% - 97.7% [85] [86] | Detection in clinical samples |
| Specificity | Gold Standard Reference | 66% - 100% [85] [86] | Detection in clinical samples |
| Limit of Detection (LoD) | Varies by kit | ~2.1 × 10² copies/µL (Animal samples) [87] | SARS-CoV-2 N gene detection |
| Cost per Reaction | Higher | ~$2 - $6 [81] | Varies with RNA extraction method |
Table 2: Analytical Comparison of PCR Detection Chemistries
| Characteristic | TaqMan Probe-Based | SYBR Green-Based |
|---|---|---|
| Chemistry Principle | Sequence-specific hydrolysis probes [83] | Intercalating dye binding dsDNA [83] |
| Specificity | High (from probe) [83] | Requires melting curve analysis [81] [82] |
| Multiplexing Potential | High (multiple probes) | Limited, but feasible [81] |
| Development Cost | High (probe synthesis) [84] | Low (primers only) [82] |
| Run Cost | High | Low |
| Flexibility | Low (fixed target) | High (easy primer redesign) |
A key study developing a multiplex SYBR Green assay reported a sensitivity of 93% and a specificity of 97% compared to a commercial Sansure Biotech kit [81]. Another in-house SYBR Green protocol demonstrated a sensitivity of 97.7% and specificity of 100% against a commercial Altona Diagnostics test [85]. The slightly variable specificity of SYBR Green methods underscores the necessity of post-amplification melting curve analysis to distinguish specific amplicons from primer-dimers or non-specific products [81] [82] [86].
Experimental Protocols
Protocol A: In-House SYBR Green One-Step Multiplex RT-PCR
This protocol is adapted from a validated method for detecting SARS-CoV-2 infection using a one-step multiplex approach [81].
3.1.1 Research Reagent Solutions Table 3: Essential Reagents for In-House SYBR Green Assay
| Reagent/Solution | Function | Example/Comment |
|---|---|---|
| Primers (N, E, β-actin) | Target-specific amplification | In-house designed; validated for specificity and absence of secondary structures [81] |
| SYBR Green One-Step Master Mix | Contains reverse transcriptase, Hot Start DNA polymerase, SYBR Green dye, dNTPs | SensiFAST SYBR No-ROX One-Step Kit [82] |
| RNA Template | Sample nucleic acid | Use crude or column-purified RNA [81] |
| Nuclease-free Water | Reaction component | To make up final volume |
3.1.2 Procedure
- Primer Design and Validation: Design primers targeting conserved regions of SARS-CoV-2 genes (e.g., N, E) and a human housekeeping gene (e.g., β-actin) as an internal control. Validate specificity using in-silico tools like Primer-BLAST and check for secondary structures [81] [82].
- Reaction Setup: Prepare a 20 µL reaction mix on ice:
- 10 µL of 2x SYBR Green One-Step Master Mix
- 0.6 µL of primer mix (e.g., 0.25 µM final concentration of each primer) [82]
- 3.8 µL of nuclease-free water
- 5 µL of extracted RNA template
- Thermal Cycling: Run the reaction on a real-time PCR instrument with the following cycling conditions:
- Reverse Transcription: 45°C for 10 min
- Enzyme Activation: 95°C for 2 min
- Amplification (40-45 cycles):
- Denaturation: 95°C for 5 s
- Annealing/Extension: 60°C for 20 s [82]
- Fluorescence acquisition at the end of this step
- Melting Curve Analysis: After amplification, generate a melting curve by:
- 95°C for 15 s
- 60°C for 1 min
- Gradual increase to 95°C (e.g., 0.3°C per second) with continuous fluorescence acquisition [81].
- Result Interpretation: Analyze the melting curve peaks. Specific amplicons will display distinct, reproducible melting temperatures (Tm), allowing differentiation from non-specific products [81].
Protocol B: Evaluation Using Commercial TaqMan Kit
This protocol outlines the procedure for using a commercial kit as a reference method for validation.
3.2.1 Procedure
- RNA Extraction: Extract RNA from nasopharyngeal/oropharyngeal swab samples using a magnetic bead-based viral nucleic acid extraction kit, following the manufacturer's instructions [19] [82].
- Reaction Setup: Prepare the reaction mix as specified in the kit's instructions (e.g., Sansure Biotech or equivalent) [81] [82]. A typical 20 µL reaction may contain:
- 9 µL of master mix
- 1 µL of primer/probe mix
- 5 µL of nuclease-free water
- 5 µL of RNA template
- Thermal Cycling: Run the reaction with conditions recommended by the manufacturer, for example:
- Reverse Transcription: 50°C for 20 min
- Enzyme Activation: 95°C for 3 min
- Amplification (40 cycles):
- Denaturation: 94°C for 10 s
- Annealing/Extension: 55°C for 30-40 s [82]
- Result Interpretation: A sample is considered positive if the Cycle Threshold (Ct) values for the target genes (e.g., ORF1ab and N) are ≤ 40 [82].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for SARS-CoV-2 PCR Detection
| Item | Function/Description | Example Brands/Types |
|---|---|---|
| Real-Time PCR Instrument | Platform for amplification and fluorescence detection | BIORAD CFX96 Touch, ABI 7500 [19] [82] |
| RNA Extraction Kit | Purification of viral RNA from clinical samples | QIAamp Viral RNA Mini Kit, apsLABS Viral Nucleic Acid Extraction Kit [19] [84] |
| Commercial TaqMan Kit | Gold standard for detection | Sansure Biotech, Roche cobas SARS-CoV-2 Test [81] [84] |
| SYBR Green Master Mix | Core reagent for in-house assays | SensiFAST SYBR No-ROX One-Step Kit [82] |
| Validated Primer Sets | For specific target amplification | WHO-recommended sets (e.g., N1, N3, ORF1ab) [84] |
| Nuclease-Free Water | Solvent for molecular reactions | Various molecular biology grades |
Application in a Broader Research Context: The Nested PCR Framework
The principles of optimizing in-house SYBR Green assays directly inform the development of more complex molecular assays, such as nested PCR for SARS-CoV-2 detection. Nested PCR significantly enhances sensitivity and specificity by using two sets of primers in sequential amplification rounds [32]. The high reproducibility (inter-assay CV < 2%) demonstrated by optimized SYBR Green assays [87] is a critical prerequisite for the robust performance of nested PCR. Furthermore, the sample preparation and RNA extraction methods validated for SYBR Green RT-PCR are directly applicable to nested PCR workflows, especially when dealing with challenging samples with low viral loads [32]. The one-step single-tube nested quantitative RT-PCR (OSN-qRT-PCR) has been shown to produce the most satisfactory results and the highest sensitivity for samples with low viral loads, such as in wastewater surveillance [32].
Both commercial TaqMan kits and in-house SYBR Green methods are effective for the molecular detection of SARS-CoV-2. The choice between them depends on the specific context of the laboratory. Commercial kits offer a turnkey solution with high reliability, making them suitable for high-throughput clinical diagnostics. In contrast, in-house SYBR Green methods provide a flexible, cost-effective alternative that, with careful optimization and validation, can achieve comparable performance. This makes them particularly valuable for research applications, resource-limited settings, and as a foundation for developing advanced assays like nested PCR. The protocols and data presented herein provide a roadmap for scientists to implement, validate, and utilize these critical molecular tools effectively.
The emergence of SARS-CoV-2 Variants of Concern (VOCs)—including Alpha (B.1.1.7), Beta (B.1.351), and Delta (B.1.617.2)—has presented significant challenges to diagnostic accuracy throughout the COVID-19 pandemic. These variants accumulated mutations in key genomic regions, potentially compromising detection efficacy in molecular assays. Nested PCR, a technique employing two successive rounds of amplification with two primer sets, offers enhanced sensitivity and specificity for viral detection, particularly advantageous for samples with low viral loads or when targeting conserved genomic regions amid viral evolution [4]. This Application Note provides a comprehensive evaluation of nested PCR performance against major VOCs and detailed protocols to ensure robust detection capabilities in research and public health settings.
Variant of Concern Profiles and Diagnostic Implications
The World Health Organization (WHO) designated Alpha, Beta, and Delta as VOCs due to their impact on transmissibility, disease severity, and potential to evade public health measures [88]. Understanding their defining mutations is crucial for diagnostic assay design.
Table 1: Characteristics of Key SARS-CoV-2 Variants of Concern
| Variant (WHO Label) | Pango Lineage | Key Spike Protein Mutations | First Detected | Impact on Severity |
|---|---|---|---|---|
| Alpha | B.1.1.7 | N501Y, D614G, P681H | September 2020 | Increased [89] |
| Beta | B.1.351 | N501Y, E484K, K417N | May 2020 | Evidence of increased virulence [90] |
| Delta | B.1.617.2 | L452R, T478K, P681R | October 2020 | Increased compared to prior variants [91] [88] |
These VOCs, particularly Delta, demonstrated significantly increased transmissibility and virulence compared to earlier lineages. The Delta variant, noted for its high transmissibility and greater virulence, became the dominant variant worldwide from mid-2021 until it was displaced by Omicron [91] [88]. The N501Y mutation, common to both Alpha and Beta variants, is associated with increased binding affinity to the human ACE2 receptor, while the L452R mutation in the Delta variant has been linked to improved viral fitness and potential antibody evasion [89] [88].
Nested PCR Assay Design for VOC Detection
Primer Design and Target Selection
Effective nested PCR assays for VOC detection must target highly conserved genomic regions to ensure robustness against mutations. The nucleocapsid (N) gene is a preferred target due to its relative stability and higher conservation compared to the spike (S) gene [4]. One validated assay targets a 633-bp segment in the first round and a 268-bp internal fragment in the second round of amplification [4].
When designing primers:
- Perform multiple sequence alignments of all major VOCs to identify conserved regions
- Avoid primer binding sites overlapping with known VOC mutation hotspots
- Validate specificity in silico against circulating variants before laboratory validation
Comparative Analytical Performance
Nested PCR demonstrates particular utility for detecting low viral loads across VOCs, with performance characteristics making it suitable for surveillance applications.
Table 2: Performance Metrics of Molecular Detection Methods for SARS-CoV-2
| Assay Type | Sensitivity | Specificity | Limit of Detection (LoD) | Key Applications |
|---|---|---|---|---|
| Nested PCR | 95% [4] | 100% [4] | ~50 copies/μL (Ct ~31.5) [4] | Detection in low viral load samples, animal surveillance [4] |
| Conventional RT-PCR | Varies by assay | Varies by assay | Generally 100-1000 copies/mL | Clinical diagnosis, widespread testing |
| Direct RT-PCR (Tata MD) | 93.9% [19] | 100% [19] | Comparable to conventional RT-PCR in lower Ct ranges [19] | Rapid, cost-effective clinical testing |
The 95% sensitivity and 100% specificity of nested PCR for SARS-CoV-2 detection, including in animal samples, highlights its reliability. Its LoD of approximately 50 copies/μL enables detection of infections with low viral loads, which is particularly valuable for surveillance studies and detecting infections in partially immune populations [4].
Detailed Experimental Protocol for Nested PCR Detection of VOCs
Sample Collection and RNA Extraction
Materials:
- Viral transport media (VTM)
- Oropharyngeal/nasopharyngeal swabs
- Commercial viral RNA extraction kit (e.g., apsLABS Viral Nucleic Acid Extraction Kit)
- Nuclease-free water
- Microcentrifuges and appropriate pipettes
Procedure:
- Collect oropharyngeal/nasopharyngeal swabs and place in VTM
- Transport to laboratory under refrigerated conditions (2-8°C) within 2 hours of collection
- Extract RNA using magnetic bead-based purification methods according to manufacturer protocols
- Elute RNA in 50-100 μL nuclease-free water
- Store extracted RNA at -80°C if not used immediately
Nested PCR Amplification
Reagents:
- Reverse transcriptase enzyme
- PCR buffer (with MgCl₂)
- dNTP mix
- Outer primer pair (specific to target gene)
- Inner primer pair (nested within first amplicon)
- DNA polymerase
- Nuclease-free water
- Positive control (SARS-CoV-2 RNA)
- Negative control (nuclease-free water)
First Round PCR Protocol:
- Prepare master mix for reverse transcription and first amplification:
- 5 μL RNA template
- 5 pmol/μL each outer primer
- 1X PCR buffer
- 200 μM dNTPs
- 1 U/μL reverse transcriptase
- 1 U/μL DNA polymerase
- Nuclease-free water to 25 μL total volume
- Run thermocycling program:
- Reverse transcription: 50°C for 30 minutes
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: 49°C for 30 seconds
- Extension: 72°C for 45 seconds
- Final extension: 72°C for 7 minutes
- Hold at 4°C
Second Round PCR Protocol:
- Prepare master mix:
- 2 μL first-round PCR product
- 5 pmol/μL each inner primer
- 1X PCR buffer
- 200 μM dNTPs
- 1 U/μL DNA polymerase
- Nuclease-free water to 25 μL total volume
- Run thermocycling program:
- Initial denaturation: 95°C for 5 minutes
- 35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: 51°C for 30 seconds
- Extension: 72°C for 30 seconds
- Final extension: 72°C for 7 minutes
- Hold at 4°C
Analysis and Interpretation
- Analyze 10 μL of second-round PCR product by gel electrophoresis (1.5-2% agarose)
- Visualize using UV transillumination after ethidium bromide or SYBR Safe staining
- Confirm expected band sizes (e.g., 268 bp for N gene target)
- Sequence positive amplicons for VOC identification if required
Diagram Title: Nested PCR Workflow for SARS-CoV-2 VOC Detection
Research Reagent Solutions
Table 3: Essential Research Reagents for Nested PCR Detection of SARS-CoV-2 VOCs
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Viral Nucleic Acid Extraction Kit (e.g., apsLABS Viral Nucleic Acid Extraction Kit) | Isolation of high-quality RNA from clinical samples | Magnetic bead-based purification preferred for consistency and yield [4] |
| N Gene-specific Primers (outer and inner sets) | Target amplification of conserved viral genomic region | N gene is more conserved with fewer mutations; higher amino acid homology [4] |
| Reverse Transcriptase Enzyme | cDNA synthesis from viral RNA | Combined with PCR components in one-step RT-PCR approach |
| DNA Polymerase | DNA amplification | Thermostable polymerase essential for PCR fidelity |
| dNTP Mix | Building blocks for DNA synthesis | Quality critical for efficient amplification |
| Agarose Gel Electrophoresis System | Amplicon visualization and size verification | Confirm expected band sizes (e.g., 268 bp for N gene) |
Discussion and Technical Considerations
The nested PCR protocol detailed herein provides a robust, cost-effective method for detecting SARS-CoV-2 VOCs, with particular utility in surveillance applications and resource-limited settings. Several factors require consideration for optimal implementation:
Variant Evolution and Primer Design: Continuous monitoring of circulating strains is essential. While the N gene is relatively conserved, ongoing evolution necessitates periodic in silico re-evaluation of primer binding sites against current variants. The approach of targeting ultra-conserved elements (UCEs) within the SARS-CoV-2 genome, as demonstrated in some RT-PCR assays, offers a strategy that may remain effective despite viral evolution [92].
Quality Control: Include appropriate controls in each run:
- Positive control: SARS-CoV-2 RNA of known concentration
- Negative control: Nuclease-free water
- Extraction control: Monitor potential contamination during RNA extraction
- Inhibition control: Ensure sample quality does not inhibit amplification
Limitations and Alternatives: While nested PCR offers high sensitivity, it carries increased contamination risk due to tube handling between amplification rounds. In clinical settings requiring rapid results, direct RT-PCR methods like the Tata MD CHECK RT-PCR XF kit that eliminate RNA extraction can provide faster turnaround (approximately one hour) while maintaining high sensitivity (93.9%) and specificity (100%) [19]. For high-throughput variant screening, variant-specific RT-PCR assays targeting key mutations can provide rapid identification without sequencing [58].
Nested PCR remains a valuable tool for SARS-CoV-2 detection and surveillance, particularly for samples with low viral loads and in research settings prioritizing cost-effectiveness. Its high sensitivity and specificity for VOCs, including Alpha, Beta, and Delta, make it suitable for large-scale animal surveillance [4] and population studies where resource constraints preclude more expensive methodologies. As SARS-CoV-2 continues to evolve, maintaining diagnostic efficacy requires ongoing primer evaluation against emerging variants and complementary use of other molecular techniques when variant identification is clinically crucial.
In the landscape of molecular diagnostics, the pursuit of sensitivity, specificity, and accessibility remains paramount. While real-time quantitative PCR (qPCR) has emerged as the conventional workhorse for pathogen detection, particularly during the SARS-CoV-2 pandemic, its limitations in detecting low viral loads have prompted the scientific community to refine alternative methodologies [80]. Among these, nested polymerase chain reaction (nested PCR) has re-established itself as a powerful technique that complements and enhances existing diagnostic frameworks. This assay operates through a two-stage amplification process utilizing two sets of primers, where the product of the first PCR reaction becomes the template for a second amplification with internal primers, thereby dramatically increasing both sensitivity and specificity [4]. This technical note explores the role of nested PCR within a broader research context, detailing its applications, protocols, and comparative advantages with a specific focus on SARS-CoV-2 detection.
Enhanced Sensitivity for Low Viral Load Detection
Addressing the False-Negative Challenge
A significant challenge in molecular diagnostics, especially during the early stages of infection or in certain sample types, is the prevalence of false-negative results from conventional PCR methods. Research has demonstrated that nested PCR effectively addresses this limitation through its dual-amplification design.
A critical study evaluating false-negative rates found that among 184 clinical samples that tested negative for SARS-CoV-2 via real-time PCR, nested PCR identified 27 positive cases (14.6%) when targeting the Orf1ab gene, 7 positive cases (3.8%) for the N gene, and 4 positive cases (2.1%) for the RdRp gene [80]. This demonstrates nested PCR's superior capability to detect viral presence in samples with low pathogen concentrations that would otherwise be missed by standard testing protocols.
Comparative Analytical Sensitivity
The enhanced sensitivity of nested PCR has been quantitatively established across multiple studies targeting various pathogens. The following table summarizes the limits of detection (LoD) achieved by nested PCR in comparison to other molecular detection methods:
Table 1: Comparative Sensitivity of Molecular Detection Methods
| Application | Method | Limit of Detection (LoD) | Reference |
|---|---|---|---|
| SARS-CoV-2 detection | Nested PCR | 0.015 ng/μL RNA | [1] |
| SARS-CoV-2 detection | Nested PCR | ~50 copies/μL (Ct 31.5) | [4] |
| SARS-CoV-2 detection | Single-round qPCR | >10 copies/μL | [27] |
| Fusarium tricinctum detection | Nested PCR | 3.1 fg/μL | [93] |
| Fusarium tricinctum detection | qPCR | 3.1 fg/μL | [93] |
| AHPND detection | Nested PCR | 1.77 copies/μL | [94] |
| AHPND detection | Conventional PCR | 177 copies/μL | [94] |
The data reveal that nested PCR consistently achieves detection limits comparable to or surpassing qPCR across diverse applications, enabling identification of pathogens at exceptionally low concentrations that challenge conventional PCR methods [27] [94].
Applications in SARS-CoV-2 Research and Beyond
SARS-CoV-2 and Variant Surveillance
The nested PCR platform has proven particularly valuable in pandemic response scenarios. Multiple research groups have developed assays targeting various regions of the SARS-CoV-2 genome, including the N gene, ORF1ab, and RdRp [1] [95] [80]. One study designed a nested PCR targeting the N gene that demonstrated 100% sensitivity and specificity when validated against approved assays, successfully detecting SARS-CoV-2 in feline samples during the first COVID-19 wave in Bulgaria [1].
Additionally, the adaptability of nested PCR has facilitated surveillance of emerging variants. Research from Indonesia developed an in-house nested PCR primer targeting ORF1ab and spike protein genes, enabling not only detection but also identification of specific mutations including T3187C, T2889C/T, G3189T (spike), and C364T (ORF1ab) through sequencing analysis [95].
Zoonotic Transmission and Animal Surveillance
The utility of nested PCR extends beyond human diagnostics to zoonotic transmission studies. As SARS-CoV-2 can infect various animal species, creating potential reservoirs for viral evolution and re-transmission to humans, surveillance in animal populations becomes crucial [4]. Nested PCR offers a cost-effective solution for large-scale animal sampling, enabling researchers to monitor viral circulation in diverse species without the prohibitive costs associated with high-throughput qPCR [4]. This application proved valuable in detecting SARS-CoV-2 in cats and dogs with respiratory symptoms, supporting One Health approaches to pandemic management [1] [4].
Broad Diagnostic Applications
The versatility of nested PCR is evidenced by its successful implementation across diverse fields:
- Acute leukemia diagnostics: Nested PCR has been established as a gold standard for detecting gene fusions in acute leukemias, though recent comparisons show qPCR may offer superior sensitivity for some targets [14].
- Plant pathogen detection: In agricultural applications, nested PCR enabled early diagnosis of Fusarium tricinctum, a fungal pathogen affecting Zanthoxylum bungeanum, with sensitivity matching qPCR [93].
- Aquaculture diagnostics: For acute hepatopancreatic necrosis disease (AHPND) in shrimp, nested PCR demonstrated exceptional sensitivity (1.77 copies/μL), significantly outperforming conventional PCR methods [94].
Comparative Methodological Analysis
Performance Metrics Across Detection Platforms
Understanding the relative strengths of different molecular detection methods is essential for appropriate platform selection. The following table provides a comparative analysis of key performance metrics:
Table 2: Performance Comparison of Molecular Detection Methods
| Parameter | Nested PCR | Conventional PCR | qPCR | LAMP |
|---|---|---|---|---|
| Sensitivity | Very High (capable of single-copy detection) | Moderate | High | High |
| Specificity | Very High (dual primer recognition) | Moderate | High | High |
| Quantification | No (endpoint detection) | No | Yes | Semi-quantitative |
| Speed | Moderate (2-3 hours) | Fast (1-2 hours) | Fast (1-2 hours) | Very Fast (30-90 min) |
| Cost per sample | Low | Low | High | Moderate |
| Equipment requirements | Standard thermocycler | Standard thermocycler | Real-time PCR system | Water bath/heat block |
| Throughput | High | High | High | Moderate |
| Risk of contamination | High (tube opening) | Low | Low | Low |
| Ease of use | Moderate (technical skill required) | Easy | Easy | Easy |
This comparative analysis reveals that nested PCR occupies a unique niche, offering maximum sensitivity and specificity while maintaining low costs, though with increased contamination risk and procedural complexity [1] [93] [4].
Experimental Protocols and Reagent Solutions
SARS-CoV-2 Nested PCR Protocol (N Gene Target)
The following protocol adapts methodologies from multiple studies for SARS-CoV-2 detection targeting the conserved N gene [1] [4]:
Primer Design and Preparation
Table 3: Primer Sequences for SARS-CoV-2 N Gene Detection
| Primer Name | Sequence (5'→3') | Position | Amplicon Size |
|---|---|---|---|
| Ext2019nCorVF | GGCAGTAACCAGAATGGAGA | 28346-28365 | 335 bp |
| Ext2019nCorVR | CTCAGTTGCAACCCATATGAT | 28681-28661 | 335 bp |
| intF | CACCGCTCTCACTCAACAT | 28432-28450 | 212 bp |
| intR | CATAGGGAAGTCCAGCTTCT | 28643-28624 | 212 bp |
RNA Extraction and cDNA Synthesis
- RNA Extraction: Extract viral RNA from 200 μL of nasopharyngeal swab samples using the ISOLATE II RNA Mini kit (Bioline) or equivalent silica membrane-based method. Elute in 50-60 μL of nuclease-free water [1] [80].
- cDNA Synthesis: Perform reverse transcription using the SensiFAST cDNA Synthesis Kit (Bioline):
- Combine 7 μL extracted RNA, 8 μL DEPC-treated water, 4 μL TransAmp buffer, and 1 μL reverse transcriptase enzyme
- Incubate at 25°C for 10 min, 42°C for 15 min, 80°C for 5 min
- Store synthesized cDNA at -20°C for immediate use or -80°C for long-term storage [1]
First Round PCR Amplification
Reaction Setup:
- 12.5 μL My Taq HS Red Mix (2×)
- 4 μL cDNA template
- 1 μL each external primer (Ext2019nCorVF/VR, 10 pmol/μL each)
- 6.5 μL PCR-grade water
- Total volume: 25 μL
Thermocycling Conditions:
Second Round (Nested) PCR Amplification
Reaction Setup:
- 12.5 μL My Taq HS Red Mix (2×)
- 0.5-1 μL first-round PCR product (template)
- 1 μL each internal primer (intF/intR, 10 pmol/μL each)
- 10-10.5 μL PCR-grade water
- Total volume: 25 μL
Thermocycling Conditions:
- Use identical parameters to first-round PCR [1]
Product Analysis
Agarose Gel Electrophoresis:
Sequencing Confirmation (Optional):
- Purify PCR products using Exo-CIP Rapid PCR Cleanup Kit
- Perform Sanger sequencing using internal primers
- Analyze sequences using BLAST against reference database (e.g., MN908947.3) [1]
Research Reagent Solutions
Table 4: Essential Research Reagents for Nested PCR Applications
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| RNA Extraction Kits | ISOLATE II RNA Mini Kit (Bioline), QIAamp Viral RNA Mini Kit (Qiagen) | Nucleic acid purification from clinical samples |
| cDNA Synthesis Kits | SensiFAST cDNA Synthesis Kit (Bioline), High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) | Reverse transcription of RNA to cDNA |
| PCR Master Mixes | My Taq HS Red Mix (Bioline), Platinum SuperFi II PCR Master Mix (Thermo Fisher) | Provides optimized buffer, enzymes, and dNTPs for amplification |
| Primer Sets | Custom-designed oligonucleotides targeting N gene, ORF1ab, RdRp | Sequence-specific amplification of target regions |
| Agarose Gel Electrophoresis | Lonza agarose, ethidium bromide/SYBR Safe, 1× TAE buffer, DNA ladders | Product separation and visualization |
| Nucleic Acid Quantification | NanoDrop 2000 (Thermo Scientific) | RNA/DNA concentration and purity assessment |
| Purification Kits | Exo-CIP Rapid PCR Cleanup Kit (New England Biolabs) | PCR product purification for sequencing |
Technical Considerations and Best Practices
Contamination Control
The two-step amplification process in nested PCR significantly increases vulnerability to contamination from amplicon carryover or cross-contamination between samples. Implementation of rigorous contamination control measures is essential:
- Physical Separation: Perform reagent preparation, sample processing, first-round PCR, second-round PCR, and product analysis in physically separated areas with dedicated equipment [27]
- Workflow Direction: Maintain unidirectional workflow from pre-amplification to post-amplification areas without backtracking
- Aerosol Prevention: Use filter tips for all liquid handling steps and positive displacement pipettes when possible
- Negative Controls: Include multiple negative controls (no-template controls, extraction controls) in each run to monitor contamination [1] [80]
- UV Irradiation: Expose workstations to UV light between procedures to degrade contaminating DNA
- Chemical Decontamination: Incorporate uracil-N-glycosylase (UNG) systems or similar enzymatic methods to degrade carryover amplicons
Primer Design and Optimization
Effective primer design is critical for successful nested PCR applications:
- Target Selection: Choose conserved genomic regions with minimal mutation rates (e.g., N gene in SARS-CoV-2) to ensure detection of variants [4]
- Specificity Verification: Validate primer specificity using in silico tools (NCBI Primer-BLAST) against relevant sequence databases
- Amplicon Sizing: Design first-round amplicons of 300-500 bp and nested amplicons of 150-250 bp for optimal efficiency
- Avoid Complementarity: Ensure primers lack self-complementarity, cross-dimers, or significant hairpin formation
- Empirical Optimization: Systematically optimize annealing temperatures and magnesium concentrations for each primer set
Nested PCR remains an indispensable tool in the molecular diagnostics arsenal, offering unparalleled sensitivity and specificity that complements and enhances conventional PCR methodologies. Its capacity to detect low pathogen loads, adaptability to diverse research applications, and cost-effectiveness position it as a valuable technique, particularly in resource-limited settings or when investigating emerging pathogens with low viral titers. While the technique demands rigorous contamination control and technical expertise, its implementation provides researchers with a powerful means to address the critical challenge of false-negative results in diagnostic testing. As molecular diagnostics continue to evolve, nested PCR maintains its relevance as a gold standard technique that bridges conventional PCR and advanced real-time methodologies, offering a balanced approach to sensitive, specific, and accessible pathogen detection.
Conclusion
Nested PCR has firmly established itself as a highly sensitive, specific, and versatile tool for SARS-CoV-2 detection, proving particularly invaluable for samples with low viral loads, complex matrices like wastewater, and in zoonotic research. Its superior performance, as validated against ddPCR and commercial RT-PCR kits, underscores its critical role in accurate diagnosis and public health surveillance. The future of this technology lies in its adaptability; ongoing primer redesign will be essential to keep pace with evolving viral variants. Furthermore, its application in wastewater-based epidemiology offers a powerful, cost-effective strategy for early community outbreak detection and real-time variant tracking, making it an indispensable asset for ongoing biomedical research and pandemic preparedness.
References
- 1. Development of Nested PCR for SARS-CoV-2 Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9231004/]
- 2. Semi-nested RT-PCR enables sensitive and high-throughput ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9052777/]
- 3. Development of a Novel Nested–RT–LAMP Assay for ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11993314/]
- 4. Nested PCR effective to detect low viral loads of SARS ... [https://www.sciencedirect.com/science/article/abs/pii/S0167587724001892]
- 5. A gamepad-like nucleic acid testing device for rapid ... [https://www.nature.com/articles/s44172-024-00229-w]
- 6. Diagnostic power of one-step and two-step RT-qPCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9152310/]
- 7. Development of highly sensitive one-step reverse ... [https://www.sciencedirect.com/science/article/pii/S0048969723064719]
- 8. Evaluation and comparison of one-step real-time PCR and ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-09574-9]
- 9. Diagnosing the novel SARS-CoV-2 by quantitative RT-PCR: variations and opportunities - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7567654/]
- 10. Nested PCR effective to detect low viral loads of SARS-CoV-2 in animal samples - PubMed [https://pubmed.ncbi.nlm.nih.gov/39128181/]
- 11. Analysis and validation of a highly sensitive one-step nested quantitative real-time polymerase chain reaction assay for specific detection of severe acute respiratory syndrome coronavirus 2 | Virology Journal | Full Text [https://virologyj.biomedcentral.com/articles/10.1186/s12985-020-01467-y]
- 12. Comparison of RT-PCR, RT-nested PCRs, and real-time ... [https://www.nature.com/articles/s41598-021-96066-4]
- 13. Dynamics of SARS-CoV-2 Mutations in Wastewater ... [https://www.mdpi.com/1422-0067/26/9/4324]
- 14. Nested-PCR vs. RT-qPCR: A Sensitivity Comparison in the ... [https://www.mdpi.com/2673-8856/4/3/19]
- 15. Diminished amplification of SARS-CoV-2 ORF1ab in a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8634734/]
- 16. Structural stabilization of the intrinsically disordered SARS ... [https://www.nature.com/articles/s41467-025-61861-4]
- 17. Development of Nested PCR for SARS-CoV-2 Detection ... [https://www.mdpi.com/2306-7381/9/6/272]
- 18. SARS-CoV-2 ORF3a drives dynamic dense body formation ... [https://www.nature.com/articles/s41467-025-59475-x]
- 19. Optimizing SARS-CoV-2 Detection: A Rapid, Cost-Effective ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12374254/]
- 20. SARS-CoV-2-specific humoral immunity in a Norwegian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12135409/]
- 21. Seroprevalence and molecular detection of SARS-CoV-2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12619265/]
- 22. Humoral response against COVID-19 in the population of ... [https://www.frontiersin.org/journals/public-health/articles/10.3389/fpubh.2025.1648937/full]
- 23. Seroprevalence of SARS-CoV-2 and humoral immune ... [https://harmreductionjournal.biomedcentral.com/articles/10.1186/s12954-024-01023-9]
- 24. Novel method for the identification of circulating SARS-CoV-2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12623389/]
- 25. Structure-Based Primer Design Minimizes the Risk of PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8573093/]
- 26. SARS‐CoV‐2 variant with mutations in N gene affecting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8661661/]
- 27. Sensitive and Quantitative Detection of Severe Acute ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7107825/]
- 28. Classification and specific primer design for accurate ... [https://www.nature.com/articles/s41598-020-80363-5]
- 29. CoVrimer: A tool for aligning SARS-CoV-2 primer ... [https://www.sciencedirect.com/science/article/pii/S0888754321002986]
- 30. Analysis and validation of a highly sensitive one-step nested ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7768088/]
- 31. Comparison of ordinary reverse transcription real-time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10482794/]
- 32. Effective sample preparation and improved RT-PCR for ... [https://www.sciencedirect.com/science/article/abs/pii/S1438463922001006]
- 33. RT-qPCR half-reaction optimization for the detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8687447/]
- 34. M gene targeted qRT-PCR approach for SARS-CoV-2 virus ... [https://www.nature.com/articles/s41598-023-43204-9]
- 35. Analytical sensitivity and efficiency comparisons of SARS ... [https://www.nature.com/articles/s41564-020-0761-6]
- 36. Comparison of analytical sensitivity of SARS-CoV-2 ... [https://www.sciencedirect.com/science/article/pii/S1201971221006767]
- 37. Optimized protocol for direct extraction of SARS-CoV-2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12051151/]
- 38. Concentration, extraction and amplification [https://www.sciencedirect.com/science/article/abs/pii/S0048969724054354]
- 39. A rapid and high-yield method for nucleic acid extraction [https://www.nature.com/articles/s41598-025-95226-0]
- 40. Optimization and comparative study of SARS-CoV-2 RNA ... [https://pubmed.ncbi.nlm.nih.gov/41135702/]
- 41. Wastewater Speaks: Evaluating SARS-CoV-2 Surveillance ... [https://www.mdpi.com/2076-2607/13/4/924]
- 42. Measuring SARS-CoV-2 RNA in Bangkok wastewater ... [https://www.nature.com/articles/s41598-025-94938-7]
- 43. SARS‐CoV‐2 Reverse Zoonosis Among Cats in China [https://pmc.ncbi.nlm.nih.gov/articles/PMC11065897/]
- 44. Seroprevalence of SARS-CoV-2 in cats from COVID-19 ... [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1542397/full]
- 45. Molecular detection, phylogenetic and whole genome ... [https://www.sciencedirect.com/science/article/abs/pii/S0034528825003121]
- 46. Publications [https://data.wastewaterscan.org/publications/]
- 47. An interpretative review of the wastewater-based ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1338100/full]
- 48. Nested Polymerase Chain Reaction - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/nested-polymerase-chain-reaction]
- 49. A novel, highly sensitive, one-tube nested quantitative real ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10714840/]
- 50. A scoping review of global SARS-CoV-2 wastewater-based ... [https://www.sciencedirect.com/science/article/pii/S2405844024066313]
- 51. Dynamics of SARS-CoV-2 Mutations in Wastewater ... [https://pubmed.ncbi.nlm.nih.gov/40362561/]
- 52. From Wastewater to Epidemiological Insights: A Systematic ... [https://www.sciencedirect.com/science/article/pii/S0043135425018809]
- 53. Semi-nested RT-PCR enables sensitive and high- ... [https://www.sciencedirect.com/science/article/abs/pii/S0009898122011263]
- 54. Evaluation of RT-PCR assays for detection of SARS-CoV-2 ... [https://www.nature.com/articles/s41598-023-28275-y]
- 55. Molecular Diagnostic Tests for SARS-CoV-2 [https://www.fda.gov/medical-devices/covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2]
- 56. Clinical performance of a dual-target SARS CoV-2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12613554/]
- 57. High capacity clinical SARS-CoV-2 molecular testing using ... [https://www.nature.com/articles/s43856-024-00531-w]
- 58. Development of new RT-PCR assays for the specific ... [https://www.sciencedirect.com/science/article/pii/S0048969724065215]
- 59. Evaluation of PCR-enhancing approaches to reduce ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969724069250]
- 60. Assessment of PCR Inhibitor Removal Methods to Monitor ... [https://link.springer.com/article/10.1007/s11270-023-06821-8]
- 61. Wastewater Based Surveillance of SARS-CoV-2: Challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8789553/]
- 62. Performance and suitability of wastewater based- ... [https://www.nature.com/articles/s41598-025-16995-2]
- 63. Identification of mutations in SARS-CoV-2 PCR primer ... [https://www.nature.com/articles/s41598-022-21953-3]
- 64. SARS-CoV-2 Mutations and Variants May Muddle the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9414863/]
- 65. New RT-PCR Assay for the Detection of Current and Future ... [https://www.mdpi.com/1999-4915/15/1/206]
- 66. A two-step process for in silico screening to assess the ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08999-3]
- 67. Underestimation of SARS-CoV-2 in wastewater due to ... [https://www.sciencedirect.com/science/article/pii/S2589914724000112]
- 68. Rapid screening for SARS-CoV-2 variants of concern in ... [https://www.sciencedirect.com/science/article/pii/S004313542100302X]
- 69. Rapid detection of the SARS-CoV-2 omicron variants ... [https://www.nature.com/articles/s41598-024-79254-w]
- 70. varVAMP: degenerate primer design for tiled full genome ... [https://www.nature.com/articles/s41467-025-60175-9]
- 71. SARS-CoV-2 - The value of Ct values [https://logicalbiological.com/sars-cov-2-the-value-of-ct-values/]
- 72. Cycle Threshold Values as Indication of Increasing SARS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10521603/]
- 73. RT-qPCR Testing and Performance Metrics in the COVID- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11394961/]
- 74. Cycle threshold of SARS-CoV-2 RT-PCR as a driver ... [https://www.nature.com/articles/s41598-024-52984-7]
- 75. Cycle Threshold (Ct) Values of SARS-CoV-2 Detected with the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10217001/]
- 76. Direct Comparison of SARS-CoV-2 Analytical Limits ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7448668/]
- 77. Comparison of Droplet Digital PCR to Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3553899/]
- 78. Evaluating the sensitivity of droplet digital PCR for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10903512/]
- 79. Comparing the precision of two digital PCR applications for ... [https://www.nature.com/articles/s41598-025-13143-8]
- 80. Evaluation of False Negative Among SARS-COV-2 ... [https://brieflands.com/journals/jjm/articles/122889]
- 81. Development and validation of cost-effective one-step ... [https://www.nature.com/articles/s41598-022-10413-7]
- 82. Development and validation of cost-effective SYBR Green ... [https://www.nature.com/articles/s41598-024-52250-w]
- 83. Comparison of SYBR Green and TaqMan methods in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3988599/]
- 84. Comparison of analytical sensitivity and efficiency for ... [https://link.springer.com/article/10.1007/s00253-022-11822-4]
- 85. Development and validation of an in-house, low-cost SARS ... [https://www.sciencedirect.com/science/article/pii/S1876034121001969]
- 86. Comparison of real-time SYBR green dengue assay with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3271430/]
- 87. Performance evaluation of a SYBR Green-Based Real ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093425001521]
- 88. Emergence of SARS-CoV-2 Delta Variant and Effect ... [https://wwwnc.cdc.gov/eid/article/29/10/23-0055_article]
- 89. SARS-CoV-2 variants of concern as of 31 October 2025 - ECDC [https://www.ecdc.europa.eu/en/covid-19/variants-concern]
- 90. Comparison of six COVID-19 serology assays for detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12282051/]
- 91. The impact of SARS-CoV-2 VOCs on clinical outcomes [https://pmc.ncbi.nlm.nih.gov/articles/PMC12393221/]
- 92. New RT-PCR Assay for the Detection of Current and Future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9863853/]
- 93. Development and comparative evaluation of LAMP, nested ... [https://pubmed.ncbi.nlm.nih.gov/40885921/]
- 94. Comparative study on nucleic acid amplification based ... [https://www.sciencedirect.com/science/article/abs/pii/S0044848625004508]
- 95. Nested polymerase chain reaction for coronavirus disease ... [https://onehealthjournal.org/Vol.10/No.2/3.php]