Virus Isolation in Cell Culture: From Foundational Principles to Advanced Applications in Biomedical Research
This comprehensive article provides researchers, scientists, and drug development professionals with an in-depth examination of cell culture methodologies for virus isolation.
Virus Isolation in Cell Culture: From Foundational Principles to Advanced Applications in Biomedical Research
Abstract
This comprehensive article provides researchers, scientists, and drug development professionals with an in-depth examination of cell culture methodologies for virus isolation. Covering both traditional and modern approaches, it explores foundational principles, practical applications across virology and vaccine development, troubleshooting for common challenges like contamination, and validation techniques for ensuring result accuracy. The content synthesizes current best practices with emerging technologies, addressing critical needs in biopharmaceutical production, diagnostic development, and therapeutic discovery while considering both technical implementation and research quality assurance.
The Evolution and Core Principles of Viral Cell Culture Systems
The evolution of cell culture represents a cornerstone of virology and biomedical research, marking a significant transition from whole-animal models to sophisticated in vitro systems. This progression has been driven by the need for more ethically acceptable, cost-effective, and physiologically relevant models for studying viral pathogenesis, developing vaccines, and screening antiviral compounds. For virus isolation research, the shift has moved from animal inoculation and embryonated eggs to two-dimensional (2D) monolayer cultures, and more recently, to three-dimensional (3D) models and organ-on-a-chip technologies [1] [2]. These advanced systems aim to closely mimic the in vivo microenvironment, providing more accurate data on viral behavior and host interactions, which is crucial for translational research and drug development [3] [4]. This application note details the key historical milestones, provides comparative data, and outlines practical protocols that trace this transformative journey.
Historical Timeline and Key Transitions
The methodology for virus isolation and culture has undergone profound changes over more than a century. The table below summarizes the major epochs in this development.
Table 1: Historical Epochs in Cell Culture for Virology
| Time Period | Primary Model | Key Advantages | Inherent Limitations |
|---|---|---|---|
| Pre-1950s | Laboratory Animals & Embryonated Eggs [1] [5] | Provided a whole-organism context for infection | High cost, ethical concerns, limited throughput, species-specific differences [6] |
| 1950s–1990s | Traditional 2D Cell Culture (Primary cells & immortalized lines) [1] [5] | Gold standard for virus isolation; cost-effective; convenient [1] [5] | Limited physiological relevance (altered polarity, morphology); does not fully replicate in vivo complexity [4] |
| 2000s–Present | Advanced 3D Cultures & Organ-on-a-Chip Models [3] [4] [7] | Mimics tissue microarchitecture and cell-cell interactions; improved pathophysiological relevance [4] [7] | Technically challenging; higher cost; lack of standardized protocols [8] [4] |
The pivotal turn towards in vitro methods began with Ross G. Harrison's 1907 demonstration of growing frog embryo tissues in clotted lymph [2]. The field was further advanced by Alexis Carrel's work on long-term cell cultivation and the introduction of antibiotics in the 1940s to prevent contamination [2]. A landmark achievement was the establishment of the first immortal human cell line, HeLa, in 1951, which revolutionized biomedical research and vaccine development, notably for polio [2]. The late 20th and early 21st centuries have been defined by innovations such as transfection, gene editing, co-culture systems, and 3D culture, collectively enabling more precise and human-relevant virology research [2].
Quantitative Comparison of Culture Models
The transition between models is justified by quantifiable differences in physiological relevance, throughput, and functional output. The following table compares the core characteristics of 2D, 3D, and perfused microfluidic cultures.
Table 2: Quantitative and Functional Comparison of Cell Culture Models
| Characteristic | 2D Static Culture | 3D Spheroid/Organoid Culture | Perfused Organ-on-a-Chip |
|---|---|---|---|
| Physiological Relevance | Low; altered cell morphology and polarity [4] | High; recapitulates tissue microarchitecture and cell-ECM interactions [4] [7] | Very High; introduces fluid shear stress and mechanical cues [8] |
| Throughput & Cost | High throughput; low cost [8] | Medium throughput; moderate cost [4] | Low throughput; high cost and complexity [8] |
| Drug Screening Concordance | Low (∼8% concordance with clinical trials in animal models highlights 2D limitations) [4] | Improved predictive value for drug efficacy and toxicity [4] [7] | High potential for predicting human pharmacokinetics and efficacy [8] |
| Specific Biomarker Expression | Baseline levels | Enhanced expression in certain contexts | Can induce specific biomarkers >2-fold (e.g., CYP3A4 in Caco-2 cells) [8] |
| Typical Applications in Virology | Routine virus isolation, plaque assays, vaccine production (e.g., FMDV in BHK-21 cells) [9] [5] | Modeling complex viral infections (e.g., respiratory viruses), host-pathogen interactions [3] | Modeling viral entry via vascular flow, systemic infection, and barrier functions (e.g., lung, intestine) [8] [3] |
A meta-analysis of perfused chip models versus static cultures found that while gains in perfusion are modest in 2D, 3D cultures show a slight improvement with flow, suggesting that high-density cell cultures benefit more from perfusion [8]. Furthermore, only specific biomarkers in certain cell types, particularly those from blood vessels, intestine, and liver, react strongly to flow conditions [8].
Established Protocols for Virus Isolation and Culture
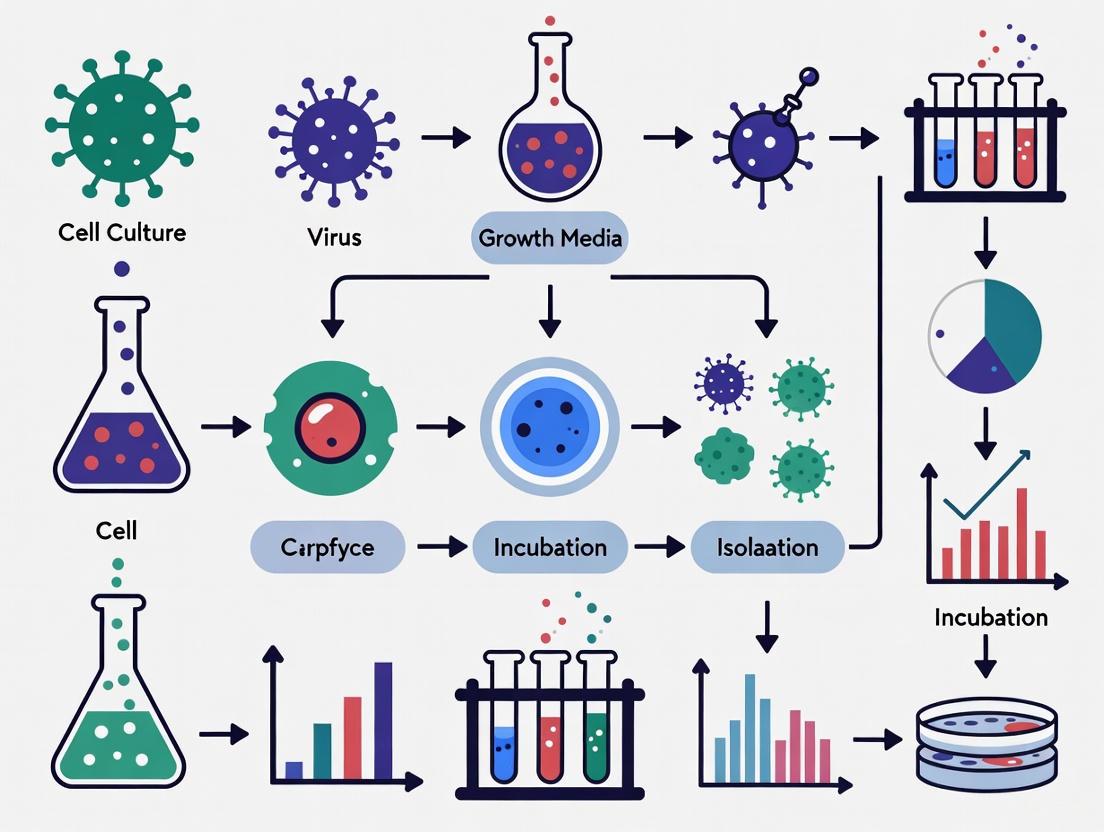
Protocol: Traditional Tube Culture for Virus Isolation
This method, the long-standing gold standard, relies on observing virus-induced cytopathic effects (CPE) on monolayer cells [1] [5].
Research Reagent Solutions:
- Cell Lines: Primary cells (e.g., Rhesus Monkey Kidney - RhMK), human diploid fibroblasts (e.g., MRC-5), or continuous lines (e.g., A549, HEp-2) [1] [5].
- Growth Medium: Eagle's Minimum Essential Medium (EMEM) or Dulbecco's Modified Eagle Medium (DMEM), supplemented with fetal bovine serum (FBS) and antibiotics [1] [10].
- Maintenance Medium: Serum-free or low-serum version of the growth medium to encourage viral replication over cell proliferation.
Methodology:
- Sample Preparation: Vortex the clinical sample (e.g., swab in transport medium) and centrifuge. Use the supernatant for inoculation [5].
- Inoculation: Aspirate growth medium from a confluent monolayer in a screw-cap tube (or shell vial) and add 0.2-0.3 mL of the processed sample inoculum [5].
- Adsorption: Incubate at 35°C with 5% CO₂ for 60-90 minutes to allow viral adsorption to cells [5].
- Maintenance: Remove the inoculum, wash the monolayer if necessary, and add maintenance medium. Return the culture to the incubator [5].
- CPE Monitoring: Examine the monolayer daily under an inverted microscope for signs of CPE (e.g., cell rounding, syncytia formation, detachment). The time to CPE appearance is virus-dependent (e.g., 1-2 days for HSV; 5-10 days for many enteroviruses; up to 30 days for CMV) [1] [5].
- Virus Identification: Confirm the isolated virus using immunofluorescence assays with virus-specific antibodies [5].
Figure 1: Workflow for traditional virus isolation via cell culture.
Protocol: Modern Rapid Culture using Shell Vials and Co-culture
This method accelerates virus detection by combining centrifugation-enhanced infection and early immunostaining, often using mixed cell lines [1] [5].
Research Reagent Solutions:
- Shell Vials: Small vials containing a monolayer on a coverslip.
- Cryopreserved Cells: Ready-to-use monolayers (e.g., MRC-5, A549, R-Mix cells) [5].
- Monoclonal Antibodies: Fluorescein-labeled (FITC) antibodies targeting specific viruses (e.g., influenza, RSV, adenovirus) or cocktail antibodies for multiple targets [5].
- Fixative: Acetone or methanol.
Methodology:
- Inoculation: Add the processed sample to a shell vial with a confluent monolayer.
- Centrifugation: Centrifuge the vial at low speed (e.g., 700 × g for 30-60 minutes) to enhance viral adsorption via low-speed centrifugation.
- Incubation: Incubate at 35°C with 5% CO₂ for 24-48 hours.
- Staining: Remove the medium, wash, and fix the cells. Add the virus-specific fluorescein-labeled antibody.
- Detection: Examine the fixed monolayer under a fluorescence microscope. Positive samples show characteristic fluorescence, allowing for diagnosis within 1-2 days [5].
Protocol: Generating 3D Spheroids using the Hanging Drop Method
3D spheroids provide a more in vivo-like model for studying virus-host interactions in a tissue-like context [4].
Research Reagent Solutions:
- Hanging Drop Plates (HDP): Commercially available plates with access holes for creating hanging drops [4].
- Cell Repellent Surface Plates: Ultra-low attachment (ULA) plates coated with poly-HEMA or commercially treated surfaces to force cell aggregation [4].
- Cell Suspension: Single-cell suspension in standard growth medium, often with reduced serum.
Methodology:
- Cell Preparation: Create a single-cell suspension and adjust the cell concentration to the desired density (e.g., 1-5 × 10⁴ cells/mL, depending on spheroid size).
- Dispensing:
- Hanging Drop Method: Pipette droplets (e.g., 20-40 µL) of cell suspension onto the lid of a petri dish. Invert the lid and place it over a dish containing PBS to maintain humidity. Cells aggregate at the bottom of the drop to form a single spheroid [4].
- ULA Plate Method: Seed the cell suspension directly into the wells of an ultra-low attachment plate. The coating prevents adhesion, forcing cells to aggregate into spheroids, often aided by gentle shaking or centrifugation [4].
- Culture: Incubate the plates at 37°C with 5% CO₂ for 24-72 hours to allow spheroid formation.
- Infection: Once mature spheroids form, introduce the virus of interest directly into the medium. Monitor for infection via reporter assays, microscopy, or metabolite analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Modern Cell Culture in Virology
| Reagent / Material | Function & Application | Example Use Case |
|---|---|---|
| Ultra-Low Attachment (ULA) Plates | Prevents cell adhesion, forcing aggregation and spheroid formation [4] | Generating uniform tumor spheroids for oncolytic virus studies |
| Chemically Defined Media (CDM) | Serum-free media with defined components; improves reproducibility and reduces contamination risk from animal sera [10] | Culturing CAR-T cells or stem cell-derived organoids for host-pathogen research |
| Hydrogels (e.g., Matrigel, Collagen) | Provides a 3D extracellular matrix (ECM) scaffold for embedded 3D culture and organoid growth [4] | Modeling respiratory virus infection in airway epithelial organoids |
| R-Mix Cells | A commercial cocultured cell line (A549 & mink lung) for isolating a broad spectrum of respiratory viruses [5] | Rapid shell vial culture for simultaneous detection of influenza, RSV, and adenovirus |
| Magnetic 3D Bioprinting Kits | Uses magnetic levitation to assemble cells into complex 3D structures for co-culture models [4] [7] | Creating a vascularized tissue model to study viral dissemination |
Signaling Pathways in Viral Entry: FMDV as a Model
The choice of cell culture system is critically influenced by the expression of specific viral entry receptors. Foot-and-mouth disease virus (FMDV) provides a clear example of how receptor usage dictates cell line susceptibility and is a key consideration in model selection [9].
Figure 2: FMDV cellular entry pathways via integrin or heparan sulfate receptors.
FMDV typically initiates infection by binding to integrin receptors (αVβ1, αVβ3, αVβ6, αVβ8) via a highly conserved RGD motif located on the VP1 capsid protein [9]. This interaction, characteristic of field viruses, triggers clathrin-mediated endocytosis. The acidic environment of the endosome then promotes capsid disassembly and release of the viral genome into the cytoplasm [9]. In contrast, cell-culture-adapted strains of FMDV often utilize heparan sulfate (HS) proteoglycans as an alternative receptor. This entry pathway occurs via caveolae-mediated endocytosis [9]. The differential expression of these receptors (e.g., high αVβ6 in epithelial cells) explains the tropism of the virus and underscores why certain cell lines (e.g., BHK-21, primary bovine thyroid cells) are selected for its isolation and propagation [9].
Cell culture serves as a fundamental tool in virology research, providing the necessary living systems for virus isolation, propagation, and pathogenesis studies [11]. These techniques are increasingly favored in pharmacological research and disease modeling due to significant advantages over animal models, including reduced costs, time constraints, and ethical concerns regarding animal use [6]. The intact animal ultimately serves as the source of all cells for culture, which can be obtained from various organs and tissues of embryonic, infant, or adult origin [6]. Cultures of animal cells are systematically classified into three distinct categories: primary cells, cell strains, and continuous cell lines, each possessing unique characteristics that determine their specific applications in virology [6] [12]. Understanding these classifications is essential for researchers investigating viral contaminants such as Epstein-Barr virus (EBV) and Ovine Herpesvirus 2 (OvHV-2), which pose significant challenges to research integrity and bioprocess safety [6].
The selection of an appropriate cell culture system directly impacts the success of viral isolation and propagation efforts. Different viruses exhibit specific tissue tropisms and require particular cellular receptors for successful infection and replication [9] [13]. For instance, foot-and-mouth disease virus (FMDV) primarily utilizes integrin receptors found on the surface of susceptible cells, with infection efficiency varying considerably between primary cells and continuous cell lines [9]. Similarly, African swine fever virus (ASFV) isolation has traditionally relied on primary macrophage cultures due to their high sensitivity, though these systems present challenges including low cell yield and contamination risks [14]. This application note provides a comprehensive comparison of primary cells, cell strains, and continuous cell lines, with detailed protocols and implementation frameworks to guide virology researchers in selecting and maintaining appropriate cell culture systems for virus isolation studies.
Classification and Characteristics of Cell Culture Systems
Comparative Analysis of Cell Culture Types
Cell culture systems are broadly categorized into three distinct types based on their origin, lifespan, and characteristics in vitro. The table below summarizes the key features, advantages, and limitations of each category:
Table 1: Characteristics of primary cells, cell strains, and cell lines
| Characteristic | Primary Cells | Cell Strains | Continuous Cell Lines |
|---|---|---|---|
| Origin | Freshly isolated from animal organs or tissues through mechanical or enzymatic methods [12] | Derived from primary cultures that have been subcultured but have not yet undergone transformation [6] | Derived from transformed cells or tumors; often immortalized [12] |
| Lifespan | Finite (usually limited to a few passages) [12] | Finite (capable of 20-80 population doublings before senescence) [6] | Infinite (can be subcultured indefinitely) [12] |
| Growth Characteristics | Exhibit anchorage dependency and contact inhibition [12] | Exhibit anchorage dependency and contact inhibition [6] | May not exhibit anchorage dependency or contact inhibition; can grow in suspension [12] |
| Genetic Profile | Diploid karyotype; genetically similar to original tissue [6] | Diploid karyotype maintained [6] | Aneuploid or heteroploid karyotype; genetically different from original tissue [12] |
| Applications | Virus isolation with high clinical relevance; vaccine production [14] [11] | Research applications requiring more material than primary cells can provide [6] | Large-scale virus propagation; high-throughput screening; basic research [9] [13] |
Cell Culture Systems in Virus Isolation
The selection of an appropriate cell culture system significantly influences the efficiency of virus isolation and propagation. Different viruses exhibit varying tropisms for specific cell types based on the presence of particular surface receptors required for viral entry [9]. For example, foot-and-mouth disease virus (FMDV) primarily utilizes integrin receptors (αVβ1, αVβ3, αVβ6, and αVβ8) found on the surface of susceptible cells, with infection efficiency varying considerably between primary cells and continuous cell lines [9]. Similarly, African swine fever virus (ASFV) isolation has traditionally relied on primary macrophage cultures due to their high sensitivity, though these systems present challenges including low cell yield and contamination risks [14].
Certain continuous cell lines have been specifically engineered or selected for enhanced susceptibility to particular viruses. For instance, the MDCK cell line is widely used for influenza virus propagation, while Vero cells are commonly employed for arbovirus isolation [13] [15]. The H1-HeLa cell line has been developed specifically for human rhinovirus 16 propagation, demonstrating how continuous cell lines can be optimized for specific virological applications [13]. Despite these advantages, primary cells often remain superior for initial virus isolation from clinical specimens due to their preserved physiological receptors and higher sensitivity to wild-type viruses [11] [15].
Table 2: Susceptible cell lines and preferred detection methods for viral contamination
| Virus | Susceptible Cell Lines | Preferred Detection Methods |
|---|---|---|
| Epstein-Barr Virus (EBV) | B95-8 [13] | PCR assays (detects active and latent forms) [6] |
| Ovine Herpesvirus 2 (OvHV-2) | Various animal and insect cell lines [6] | Specific PCR assays; cytopathic effect observation [6] |
| Foot-and-Mouth Disease Virus (FMDV) | BHK-21, IB-RS-2, ZZ-R 127, LFBKvB6, primary bovine kidney cells [9] | Virus neutralization tests; plaque assays; cytopathic effect observation [9] |
| African Swine Fever Virus (ASFV) | Primary porcine bone marrow cells, primary macrophage cultures, MA-104 [14] | Hemadsorption assay; real-time PCR; cytopathic effect observation [14] |
Experimental Protocols for Cell Culture in Virus Isolation
Protocol 1: Preparation of Porcine Bone Marrow Primary (PBMP) Cell Culture for ASFV Isolation
Porcine bone marrow primary (PBMP) cell culture offers high sensitivity for African swine fever virus (ASFV) isolation, resulting in high viral yields with minimal contamination risk [14]. This protocol adapts traditional methods to enhance cell yield and reduce contamination, addressing limitations of other primary culture systems such as blood leukocytes and alveolar macrophages.
Materials and Reagents
- Source Animals: Clinically healthy piglets (4-6 weeks old, 8-15 kg) from farms free from ASF, CSF, and other infectious diseases [14]
- Basal Medium: Minimum Essential Medium (MEM) [14]
- Supplements: Fetal Bovine Serum, Gentamicin (40mg/ml), Penicillin G, Streptomycin sulfate [14]
- Digestion Reagent: TrypLE Express Enzyme [14]
- Filter Systems: EZFlow cell strainers [14]
- Culture Vessels: T-25, T-75, and T-225 flasks [14]
Procedure
Necropsy and Bone Marrow Collection:
- Euthanize donor piglet following approved ethical guidelines
- Aseptically collect long bones (femur and tibia) and place in sterile saline solution with antibiotics
- Transfer to biological safety cabinet for all subsequent procedures [14]
Bone Marrow Extraction:
- Remove muscle tissue and cartilage from bone surfaces
- Cut bone ends to expose marrow cavity
- Flush marrow cavity with MEM supplemented with antibiotics using syringe with needle
- Collect marrow plugs in sterile centrifuge tube [14]
Cell Dissociation and Filtration:
- Dissociate bone marrow plugs by repeated pipetting
- Filter cell suspension through EZFlow cell strainer to remove bone fragments and debris
- Centrifuge filtered suspension at 400 × g for 10 minutes
- Resuspend cell pellet in complete growth medium [14]
Cell Seeding and Culture:
- Seed cells at density of 5 × 10^6 cells/cm² in appropriate culture vessels
- Maintain cultures at 37°C in 5% CO₂ humidified incubator
- Replace medium every 2-3 days until confluent monolayer forms (typically 5-7 days) [14]
Quality Control:
- Monitor cultures daily for contamination and cell morphology
- Confirm macrophage phenotype through morphological assessment
- Use only cultures with >95% viability for virus isolation [14]
Applications
PBMP cultures are specifically recommended for ASFV isolation from field samples, even with low virus loads [14]. These cultures support high viral replication and exhibit the characteristic hemadsorption phenomenon, facilitating virus identification.
Protocol 2: Isolation and Culture of Primary Human Corneal Epithelial Cells (HCECs)
Primary human corneal epithelial cells (HCECs) provide a physiologically relevant platform for therapeutic drug testing, offering significant advantages over immortalized cell lines that may exhibit altered gene expression profiles [16]. This protocol standardizes the isolation and culture process to address challenges such as low purity, variable yield, and limited passages.
Materials and Reagents
- Source Tissue: Human corneoscleral buttons from donors (age 18-70 years) with no history of ocular disease [16]
- Basal Medium: Corneal Epithelial Cell Basal Medium [16]
- Growth Supplements: Corneal Epithelial Cell Growth Kit (contains apo-transferrin, epinephrine, Extract P, hydrocortisone hemisuccinate, L-glutamine, rh insulin, CE growth factor) [16]
- Digestion Enzyme: Dispase II solution (15 mg/mL in complete growth medium) [16]
- Coating Substrate: Matrigel matrix solution [16]
- Antibiotics: Penicillin/Streptomycin solution [16]
Procedure
Corneoscleral Button Preparation:
- Transfer corneoscleral button to petri dish using forceps
- Rinse three times with cold HBSS containing 1% Penicillin/Streptomycin
- Carefully trim away remaining scleral tissue to prevent non-corneal epithelial cell contamination [16]
Epithelial Cell Isolation:
- Immerse entire corneoscleral button in 5 mL Dispase II solution (15 mg/mL)
- Incubate at 4°C for 16-24 hours to complete digestion process
- Gently separate epithelial sheet from underlying stroma using forceps
- Dissociate epithelial sheet into single cells by repeated pipetting [16]
Surface Coating:
- Coat culture flasks or plates with diluted Matrigel matrix solution
- Incubate at 37°C for at least 1 hour before cell seeding
- Remove excess coating solution before adding cell suspension [16]
Cell Seeding and Culture:
- Seed isolated HCECs onto Matrigel-coated surfaces at appropriate density
- Culture in complete growth medium with specialized supplements
- Maintain at 37°C in 5% CO₂ humidified incubator
- Change medium every 2-3 days [16]
Subculture:
- Detach cells using 0.05% Trypsin-0.02% EDTA when 80-90% confluent
- Neutralize trypsin with Trypsin-neutralizing solution
- Subculture at ratio of 1:3 to 1:4
- Note: Subsequent passages (from passage 3 onward) no longer require Matrigel coating [16]
Functional Validation
- Perform immunofluorescence staining with established markers to identify limbal stem cells and differentiated corneal epithelial cells
- Conduct Ca²⁺ assay to validate functionality by demonstrating intracellular Ca²⁺ release in response to ATP stimulation [16]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential research reagents for cell culture in virus isolation
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Basal Media | Minimum Essential Medium (MEM), Corneal Epithelial Cell Basal Medium [14] [16] | Provide nutritional foundation for cell growth and maintenance |
| Growth Supplements | Fetal Bovine Serum, Corneal Epithelial Cell Growth Kit, L-glutamine [16] [14] | Supply essential growth factors, hormones, and nutrients |
| Dissociation Reagents | TrypLE Express Enzyme, Dispase II, Trypsin-EDTA [14] [16] | Facilitate tissue dissociation and cell detachment during subculturing |
| Antibiotics/Antimycotics | Penicillin/Streptomycin, Gentamicin [16] [14] | Prevent bacterial and fungal contamination |
| Surface Coatings | Matrigel, Laminin, Collagen [16] | Provide extracellular matrix support for cell attachment and growth |
| Cell Separation | EZFlow cell strainers [14] | Remove debris and obtain single-cell suspensions |
| Buffers and Salts | Phosphate Buffered Saline (PBS), D-sorbitol, HBSS [16] [14] | Maintain physiological pH and osmolarity during procedures |
Workflow and Pathway Diagrams
Decision Framework for Cell Culture Selection in Virus Isolation
Viral Infection Pathway in Cell Culture Systems
The strategic selection of appropriate cell culture systems—whether primary cells, cell strains, or continuous cell lines—represents a critical decision point in virology research that directly impacts the success of virus isolation and propagation efforts. Primary cells offer unparalleled physiological relevance and high sensitivity for initial virus isolation from clinical specimens, particularly valuable for fastidious viruses such as African swine fever virus and newly emerging pathogens [14] [15]. Cell strains provide an intermediate solution with extended lifespan while maintaining important biological characteristics, suitable for research applications requiring more material than primary cells can provide [6]. Continuous cell lines deliver consistency, scalability, and convenience for large-scale virus production and high-throughput screening applications, despite potential limitations in physiological relevance due to genetic drift and altered characteristics [9] [13].
As viral diagnostics continue to evolve, cell culture maintains its essential role alongside modern molecular techniques, providing viable virus isolates essential for pathogenesis studies, vaccine development, and antiviral testing [11] [15]. The integration of robust quality control measures, including short tandem repeat (STR) profiling and mycoplasma testing, combined with the implementation of standardized protocols like those presented in this application note, ensures the authenticity and integrity of cell cultures used in virology research [6]. Through careful matching of cell culture systems to specific research objectives, virologists can optimize their experimental outcomes while maintaining the physiological relevance necessary for translating findings into clinical and public health applications.
Essential Laboratory Setup and Safety Considerations for Virology Work
Virology research, particularly work involving virus isolation through cell culture, requires meticulously planned laboratory environments and stringent safety protocols to ensure both scientific integrity and researcher safety. The complex nature of handling infectious agents demands specialized equipment, engineered controls, and comprehensive procedural guidelines. Within the broader context of cell culture methods for virus isolation research, this application note provides detailed guidance on establishing and maintaining a virology laboratory capable of supporting advanced research while containing potential biohazards. These foundational elements enable researchers to effectively isolate and characterize viral pathogens, from common respiratory viruses to emerging threats, using both traditional culture techniques and modern molecular approaches.
Essential Virology Laboratory Equipment
A virology laboratory requires specialized equipment to facilitate the isolation, propagation, and characterization of viral pathogens. The equipment listed in Table 1 represents core components necessary for conducting virology research safely and effectively, with particular emphasis on cell culture applications for virus isolation [17].
Table 1: Essential Equipment for Virology Research
| Equipment Category | Specific Instruments | Primary Research Applications |
|---|---|---|
| Basic Laboratory Tools | Centrifuges, pipettes, pH meters, refrigerators, freezers, microscopes, water baths [17] | General sample preparation, measurement, storage, and initial observation. |
| Specialized Virology Equipment | Biosafety cabinets, autoclaves, vortex mixers, colony counters, ELISA readers [17] | Safe handling of infectious materials, sterilization, sample mixing, and quantitation. |
| Cell Culture & Virus Propagation | CO₂ incubators, shaker water baths, inoculation chambers, adjustable microscope tables [17] | Maintaining cell lines, incubating infections, observing cytopathic effects (CPE). |
| Analytical & Diagnostic Instruments | Spectrometers, mass spectrometry benches, PCR machines, RT-qPCR equipment [17] [18] | Viral load quantification, protein analysis, and molecular detection of viral genetic material. |
Research Reagent Solutions for Virus Isolation
Successful virus isolation in cell culture relies on a suite of essential research reagents and materials. The following table details key components of the "scientist's toolkit" for virological research.
Table 2: Key Research Reagent Solutions for Virus Isolation
| Reagent/Material | Function in Virology Research |
|---|---|
| Cell Lines | Serve as host systems for viral replication; selection depends on virus tropism (e.g., Caco-2 and MRC-5 for respiratory viruses) [19]. |
| Growth Media & Sera | Provide essential nutrients to maintain cell viability and support viral propagation in culture. |
| Trypsin/EDTA | Used for detaching adherent cells for subculturing and maintaining cell lines. |
| PCR/RT-qPCR Reagents | Enable detection and quantification of viral nucleic acids from clinical samples or culture supernatants [18]. |
| Primary Antibodies | Used in immunological assays (e.g., immunostaining) to detect viral antigens in infected cells. |
| Transport Media | Preserve viral integrity in clinical specimens (e.g., serum, respiratory samples) during storage and transport [18]. |
Laboratory Design and Safety Framework
Laboratory Zoning and Workflow
Effective virology laboratory design incorporates distinct zones to separate activities by function and risk level, thereby minimizing cross-contamination and enhancing operational efficiency. A well-designed lab should include dedicated areas for: sample storage and processing, handwashing and PPE storage, nucleic acid processing and storage, rapid testing and PCR, biowaste containment, and data analysis [20]. The physical layout should facilitate a unidirectional workflow, moving from clean to dirty areas, with samples processed sequentially through receiving, preparation, analysis, and decontamination stages.
The strategic placement of safety equipment is critical within this workflow. Biosafety cabinets (BSCs) must be located within the cell culture and virus isolation zones to provide primary containment during procedures that may generate aerosols [17] [20]. Emergency equipment including eyewash stations, safety showers, and fire suppression systems should be readily accessible in multiple locations, particularly in high-risk zones [17]. Surface materials also contribute significantly to safety; non-porous, anti-microbial casework and durable epoxy countertops are recommended throughout the laboratory as they are easy to decontaminate and resist bacterial growth [20].
Biosafety Levels and Risk Assessment
Virology work must be conducted at a biosafety level (BSL) appropriate to the specific pathogen being handled, with risk assessments based on factors such as pathogenicity, transmission route, and available treatments [21]. Most diagnostic virology work with agents associated with human disease (e.g., influenza, SARS-CoV-2) requires BSL-2 containment, which includes BSCs, appropriate PPE, and controlled access [22] [21]. More hazardous pathogens require BSL-3 facilities, which incorporate additional engineering controls such as specialized ventilation (negative air pressure) and scaled-up procedural requirements [21].
Core Protocols for Virus Isolation in Cell Culture
Protocol: Cell Culture "Combo" Method for Respiratory Virus Isolation
This protocol outlines a micromethod for inoculating combinations of cell lines ("cell combos") to isolate respiratory viruses that may not be detected by standard molecular techniques, thereby reviving classical virology techniques for contemporary diagnostics [19].
Materials and Reagents
- Cell Lines: Ten selected cell lines combined into five combos of two cell lines each (e.g., Caco-2/MRC-5 combo) [19]
- Growth Media: Cell-type specific media (e.g., MEM, DMEM) supplemented with fetal bovine serum and antibiotics
- Clinical Samples: Respiratory samples (e.g., nasopharyngeal swabs, aspirates) found negative by multiplex RT-PCR panels
- Equipment: Biosafety cabinet, CO₂ incubator, inverted microscope, refrigerated centrifuge, pipettes
Experimental Workflow
The following diagram illustrates the step-by-step workflow for processing samples using the cell combo method for enhanced virus isolation.
Procedure
- Cell Combo Preparation: Culture selected cell line combinations (e.g., Caco-2/MRC-5) in 96-well microplates until they reach 80-90% confluence [19].
- Sample Preparation: Centrifuge clinical samples at low speed (2000 × g for 10 minutes) to remove debris and filter through a 0.45 μm membrane.
- Inoculation: Aspirate media from cell combos and inoculate with 50-100 μL of processed sample per well. Include negative controls (media only).
- Incubation & Monitoring: Incubate inoculated cells at 35-37°C with 5% CO₂. Examine cultures daily for 10-14 days using an inverted microscope for appearance of cytopathic effects (CPE) such as cell rounding, syncytia formation, or detachment [19] [6].
- Virus Detection & Confirmation: Harvest supernatant and cells from wells showing CPE. Detect viral genetic material using RT-PCR assays or confirm novel viruses through metagenomic sequencing [19].
- Virus Stock Preparation: Create virus stocks by propagating confirmed isolates in susceptible cell lines, then aliquot and store at -80°C.
Applications and Importance
This method is particularly valuable for investigating undiagnosed respiratory infection outbreaks and detecting emerging viruses that might be missed by targeted molecular assays. The approach successfully isolated 12 herpes simplex or varicella-zoster viruses not detected by respiratory multiplex PCR assays in a proof-of-concept study [19].
Protocol: Viral Isolation from Field-Collected Ticks
This protocol describes methods for isolating and culturing viruses from field-collected ticks, facilitating research into medically significant tick-borne pathogens like Deer tick virus (DTV) and Powassan virus (POWV) [23].
Materials and Reagents
- Ticks: Field-collected ticks, identified to species and life stage
- Cell Lines: Cultured mammalian cells amenable to tick-borne virus replication (e.g., Vero cells)
- Homogenization Buffer: Sterile phosphate-buffered saline (PBS) or specialized media containing antibiotics and antifungals
- Equipment: Biosafety cabinet, tissue homogenizer, refrigerated centrifuge, CO₂ incubator
Procedure
- Tick Preparation: Surface-sterilize ticks by immersion in 70% ethanol followed by rinsing in sterile PBS.
- Homogenization: Homogenize individual or pooled ticks in cold homogenization buffer using sterile pestles or beads.
- Clarification: Centrifuge the homogenate at high speed (10,000 × g for 10 minutes at 4°C) to remove debris.
- Filtration: Filter the supernatant through a 0.45 μm membrane.
- Inoculation: Aspirate media from cultured mammalian cells and inoculate with the filtered supernatant.
- Incubation: Adsorb for 1-2 hours at 37°C, then add fresh maintenance media and incubate at 37°C with 5% CO₂.
- Monitoring & Harvest: Monitor daily for CPE. Harvest supernatant when CPE is extensive (typically 50-80% of cells affected), then clarify by centrifugation and store aliquots at -80°C.
Safety Protocols and Best Practices
Comprehensive Safety Equipment
Virology laboratories must be equipped with multiple layers of safety equipment to protect personnel and the environment. Essential safety equipment includes [17] [20]:
- Primary Containment: Biosafety cabinets (Class II or III) for all procedures with infectious materials
- Personal Protective Equipment (PPE): Lab coats, gloves, eye protection, and respiratory protection as needed
- Decontamination Equipment: Autoclaves for sterilizing infectious waste before disposal
- Emergency Equipment: Eyewash stations, safety showers, and fire suppression systems
Administrative Controls and Training
Robust administrative controls form the foundation of laboratory safety. All laboratory personnel must receive comprehensive training in [22]:
- Biosafety Protocols: Specific procedures for handling virus and virus-infected cells
- PPE Usage: Proper selection, use, and removal of personal protective equipment
- Emergency Procedures: Response to spills, exposures, and other incidents
- Waste Management: Proper segregation and decontamination of biological waste
Principal investigators are responsible for ensuring all laboratory members read and comprehend the laboratory biosafety protocol and should provide clear documentation of safety procedures, vacation/sick leave policies, and expectations regarding work hours to promote a healthy work-life balance [22].
Establishing a virology laboratory for cell culture-based virus isolation requires meticulous planning of both physical infrastructure and operational protocols. The essential components include appropriate biosafety containment, specialized equipment for virus propagation and detection, and comprehensive safety systems. The protocols outlined herein, particularly the cell culture "combo" method for respiratory virus isolation, provide powerful tools for detecting known and emerging viral pathogens that might evade standard molecular detection methods. By integrating these specialized techniques with rigorous safety practices, researchers can create a productive laboratory environment that facilitates critical virology research while ensuring the safety of personnel and the community.
Cytopathic effect (CPE) refers to the structural changes in host cells that are caused by viral invasion [24]. When a virus induces these morphological changes, it is termed cytopathogenic [24]. The observation of CPE remains a cornerstone technique in virology, serving as a critical diagnostic tool for identifying and characterizing viral infections in cell culture [24]. For researchers investigating virus isolation, CPE provides visual evidence of viral presence and replication, offering insights into viral pathogenicity and host-cell interactions.
The underlying mechanisms of CPE involve viral hijacking of cellular machinery, often culminating in cell death. This can occur through direct lysis (dissolution) of the host cell or when the cell dies without lysis due to its inability to reproduce [24]. These changes are a necessary consequence of efficient virus replication, occurring at the expense of the host cell's viability [24]. The progression of these changes is most readily observed in cell culture, where infection can be synchronized and cells can be frequently monitored [25].
Types and Classifications of CPE
Cytopathic effects manifest in various forms, each providing characteristic clues about the infecting virus. Skilled virologists can distinguish these types even in unstained, living cultures [26]. The major CPE categories are detailed in Table 1 below.
Table 1: Common Types of Cytopathic Effects (CPE) and Associated Viruses
| CPE Type | Morphological Description | Characteristic Viruses |
|---|---|---|
| Total Destruction | Complete destruction and detachment of the host cell monolayer within days [24]. | Enteroviruses [24] [27] |
| Subtotal Destruction | Partial detachment of the cell monolayer; some cells remain attached [24]. | Togaviruses, some Picornaviruses, some Paramyxoviruses [24] [27] |
| Focal Degeneration | Localized areas of infection (foci) where cells become rounded, enlarged, and refractile [24]. | Herpesviruses, Poxviruses [24] [27] |
| Swelling and Clumping | Significant cell swelling followed by clumping into clusters before detachment [24]. | Adenoviruses [24] [27] |
| Syncytium Formation | Fusion of plasma membranes of multiple cells, creating large cells with multiple nuclei (polykaryons) [24] [26]. | Paramyxoviruses, Herpesviruses, some Coronaviruses [24] [26] |
| Foamy Degeneration | Formation of large or numerous cytoplasmic vacuoles (vacuolization) [24]. | Certain Retroviruses, Flaviviruses, Paramyxoviruses [24] [27] |
| Inclusion Bodies | Abnormal insoluble structures within the nucleus or cytoplasm; areas of viral synthesis or assembly [24] [26]. | Rabies virus (cytoplasmic), Herpesviruses (nuclear), Adenoviruses (nuclear) [24] [26] |
The rate at which CPE appears is also a diagnostically useful characteristic. A virus is considered "slow" if CPE appears after 4 to 5 days in vitro at a low multiplicity of infection (MOI), and "rapid" if it appears after 1 to 2 days under the same conditions [24].
Mechanisms of CPE Induction
The structural changes observed as CPE are the visual manifestation of profound biochemical disruptions within the infected cell. Several key mechanisms contribute to this damage.
Shutdown of Host Cell Synthesis
Many cytocidal viruses code for proteins that actively shut down host cell protein synthesis, an event incompatible with long-term cell survival [26]. This shutdown is particularly rapid and severe in infections by picornaviruses, some poxviruses, and herpesviruses [26]. Cellular RNA and DNA synthesis are typically affected as a secondary consequence.
Direct Cytopathic Effects of Viral Proteins
While viruses do not produce classic toxins, viral components can be directly toxic to the cell. For instance, viral capsid proteins, such as the adenovirus penton and fiber proteins, can be a principal cause of CPE when present in high concentrations [26]. The accumulation of viral proteins late in the replication cycle is a common pathway leading to cell damage.
Membrane Alteration and Cell Fusion
Many viruses insert viral proteins into the host cell's plasma membrane, which can alter its permeability and lead to osmotic swelling [26]. Notably, viruses like paramyxoviruses and herpesviruses produce fusion proteins that cause the plasma membranes of infected cells to fuse with adjacent uninfected cells, forming syncytia [24] [26]. This allows the virus to spread directly from cell to cell, evading host antibodies [24].
Diagram 1: Key mechanisms through which viruses induce cytopathic effects.
CPE-Based Assays for Antiviral Research
The quantifiable nature of virus-induced cell death makes CPE-based assays powerful tools for antiviral drug discovery and virology research. These assays measure viral infectivity directly by assessing the potency of compounds in inhibiting the replication of infectious viruses [28].
The CPE Inhibition Assay
This assay is suitable for high-throughput screening in a 96-well plate format [28]. It typically involves infecting a cell monolayer with a virus and then measuring cell viability in the presence or absence of antiviral compounds. A common readout involves measuring cellular ATP levels, which are present in viable cells and depleted upon cell death. A reduction in luminescence signal indicates viral-induced CPE, enabling the quantitation of antiviral efficacy [29].
Plaque Assay
The plaque assay is a more labor-intensive method that serves as a secondary assay to confirm antiviral activity [28]. It involves infecting a cell monolayer with serial dilutions of a virus sample. A semi-solid overlay medium is added to prevent uncontrolled viral spread, ensuring that infection is limited to neighboring cells. Each infectious viral particle produces a clear zone of lysed cells or CPE, known as a "plaque," which can be counted to quantify infectious viral titer [28].
Determining TCID₅₀ (Tissue Culture Infective Dose)
The TCID₅₀ is the virus dilution that reduces measured cell viability by 50% [29]. This value is critical for standardizing viral inoculums in subsequent experiments, such as potency testing of antiviral agents. To determine TCID₅₀, serial dilutions of a virus stock are added to target cells. After a specified incubation period, cell viability is measured, and the results are plotted to find the dilution that causes 50% cell death [29].
Diagram 2: Generalized workflow for a CPE-based antiviral screening assay.
Table 2: Optimized Assay Conditions for Human Coronaviruses in CPE and Plaque Assays
| Virus | Assay | Cell Line | Incubation Temperature (°C) | Incubation Time (days) |
|---|---|---|---|---|
| HCoV-OC43 | CPE | RD | 33 | 4.5 |
| HCoV-OC43 | Plaque | RD | 33 | 4.5 |
| HCoV-229E | CPE | MRC-5 | 33 | 5.5 |
| HCoV-229E | Plaque | RD | 33 | 5.5 |
| HCoV-NL63 | CPE | Vero E6 | 37 | 4 |
| HCoV-NL63 | Plaque | Vero E6 | 37 | 4 |
Source: Adapted from [28]
Application Note: Quantitative CPE Measurement Using a Luminescence Assay
Background and Principle
Traditional microscopic assessment of CPE is qualitative and time-consuming. The Viral ToxGlo Assay provides a simple, mix-and-read format that quantifies cell viability based on the measurement of cellular ATP, which is present in viable cells and depleted upon viral-induced cell death [29]. Depletion of ATP leads to a reduction in luminescence signal, enabling robust quantitation of viral-induced CPE [29].
Key Advantages
- Speed: Results are obtained just 10 minutes after reagent addition [29].
- Sensitivity: Luminescence readout provides high sensitivity suitable for screening [29].
- Quantitative: Generates numerical data (RLU) for accurate calculation of values like TCID₅₀ and EC₅₀, surpassing qualitative visual assessment [29].
- Reproducibility: The assay demonstrates excellent reproducibility [28].
Protocol: Measuring Antiviral Potency (EC₅₀)
This protocol outlines the steps to determine the concentration of an antiviral compound that provides 50% protection from viral CPE (EC₅₀).
- Cell Seeding: Plate host cells (e.g., MDCK or MRC-5) at a density of 10,000 cells per well in a 96-well white microplate with clear bottoms. Include no-cell control wells. Allow cells to attach and grow overnight at 37°C and 5% CO₂ [29].
- Compound and Virus Addition:
- Prepare serial dilutions of the antiviral compound (e.g., Ribavirin, Remdesivir) in culture media.
- Add diluted compound to the cell plate.
- Add a pre-determined dilution of virus stock that will produce optimal CPE to the test wells. To assess compound cytotoxicity, add the same dilution series to a separate set of wells without virus [29].
- Incubation: Incubate the treated cell plates at the appropriate temperature (e.g., 33°C or 37°C) and 5% CO₂ for the virus-specific duration (e.g., 3-6 days) [29].
- Viability Measurement:
- Add the Viral ToxGlo ATP detection reagent to all assay wells.
- Incubate at room temperature for 10 minutes to allow cell lysis and signal development.
- Seal the plate and read the luminescent signal on a compatible microplate reader [29].
- Data Analysis:
- Plot results as Relative Light Units (RLU) versus compound concentration using a 4-parameter logistic curve fit in analysis software.
- The EC₅₀ value is the compound concentration that rescues 50% of the virus-induced CPE. The CC₅₀ (cytotoxic concentration 50) can be determined from the wells containing compound but no virus [29].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents and Materials for CPE-Based Assays
| Item | Function/Application | Example Use Case |
|---|---|---|
| Viral ToxGlo Assay Kit | Quantitative measurement of cell viability via ATP-dependent luminescence; used to quantify CPE [29]. | High-throughput screening of antiviral compounds against HCoV-229E and Influenza A (H1N1) [29]. |
| Cell Lines: Vero E6, MRC-5, RD, MDCK | Mammalian cell lines that support the replication of specific viruses and display characteristic CPE. | Vero E6 for HCoV-NL63; MRC-5 for HCoV-229E; MDCK for Influenza A virus [29] [28]. |
| Remdesivir | Nucleoside analog antiviral drug; used as a positive control in antiviral assays [28]. | Calibration of CPE and plaque assays for human coronaviruses; EC₅₀ determination [29] [28]. |
| Ribavirin | Broad-spectrum antiviral nucleoside analog; used as a positive control [29]. | Measuring protection against CPE induced by viruses like Influenza A (H1N1) [29]. |
| 96-well & 6-well Tissue Culture Plates | Platforms for cell culture; 96-well for high-throughput CPE assays, 6-well for plaque assays [28]. | CPE assay in 96-well format; plaque assay for titer determination or confirmatory testing in 6-well format [28]. |
| SpectraMax iD5 Multi-Mode Microplate Reader | Instrument for sensitive detection of luminescence signals from viability assay kits. | Reading luminescence in the Viral ToxGlo Assay [29]. |
The observation and quantification of cytopathic effects remain fundamental techniques in diagnostic virology and antiviral research. The ability to visually identify viral infection through characteristic morphological changes in cell culture provides a powerful, direct method for virus isolation and identification. Furthermore, the translation of this visual readout into robust, quantitative assays has cemented the role of CPE in modern drug discovery pipelines. By utilizing the protocols and applications detailed in this document, researchers can effectively leverage CPE to advance our understanding of viral pathogenesis and develop novel therapeutic agents to combat emerging viral threats.
Virus isolation in cell culture remains a foundational technique in clinical virology, vital for pathogen discovery, vaccine development, and antiviral drug evaluation. Despite advancements in molecular diagnostics, the ability to isolate and propagate viruses in susceptible cell lines provides an irreplaceable tool for obtaining infectious viral stocks, conducting phenotypic characterization, and detecting unknown pathogens. The selection of appropriate cell lines is paramount, as viral tropism varies significantly, and no single cell line supports the growth of all viruses. This application note details the specific uses and performance characteristics of four critical cell lines—RhMK, MRC-5, HEp-2, and A549—in the context of a broader thesis on cell culture methods for virus isolation. We provide a consolidated reference of quantitative performance data and detailed protocols to guide researchers, scientists, and drug development professionals in optimizing their viral diagnostic and research workflows.
Comparative Susceptibility of Key Cell Lines
The effectiveness of a cell line for virus isolation is measured by its susceptibility, which dictates both the range of viruses it can detect and the efficiency of isolation. The following table summarizes the core applications and performance metrics for the four key cell lines, based on published studies.
Table 1: Viral Susceptibility and Performance of Key Cell Lines
| Cell Line | Cell Type / Origin | Primary Viral Applications | Isolation Performance and Comparative Data |
|---|---|---|---|
| RhMK (Rhesus Monkey Kidney) | Primary, epithelial | Respiratory Syncytial Virus (RSV), Influenza, Parainfluenza | RSV: CPE in 50% of cultures in 5 days, 90% in 7 days [30]. Influenza/Parainfluenza: Broadly used, but may be outperformed by other lines like MDCK or CACO-2 for influenza [31]. |
| MRC-5 | Human diploid lung fibroblast | Influenza Virus, RSV, Cytomegalovirus, Adenovirus (less susceptible) | Influenza: 18% isolation rate, comparable to MDCK cells (15%) when treated with trypsin [32]. RSV: Used in combination with RhMK and HEp-2 for maximal yield; slower CPE development than RhMK [30]. HSV: 73.6% isolation rate, less sensitive than A549 (92.5%) [33]. |
| HEp-2 | Human epithelial carcinoma | Respiratory Syncytial Virus (RSV) | RSV: 48% isolation rate, considered a benchmark for HRSV isolation [34]. Often used in combination with other cells (e.g., RhMK, MRC-5) to improve overall viral detection [30]. |
| A549 | Human lung carcinoma | Adenovirus, Herpes Simplex Virus (HSV), Respiratory Viruses | Adenovirus: 93.8% isolation rate, superior to HEK (87.0%) and CMK (47.5%) cells [33]. HSV: 92.5% isolation rate, comparable to Vero (89.0%) and superior to MRC-5 (73.6%) [33]. Can be engineered for susceptibility to other viruses (e.g., HCoV-229E) [35]. |
Detailed Experimental Protocols for Virus Isolation
Protocol 1: Isolation of Respiratory Viruses using a Combination Cell Culture System
This protocol, adapted from published methods, maximizes the recovery of common respiratory viruses like RSV and influenza by utilizing the complementary tropisms of multiple cell lines [30] [32].
Application: Isolation of Respiratory Syncytial Virus (RSV), Influenza Virus, and other respiratory pathogens. Key Cell Lines: RhMK, MRC-5, HEp-2 [30] [32].
Materials and Reagents:
- Cell Lines: Confluent monolayers of RhMK, MRC-5, and HEp-2 cells in appropriate tissue culture flasks or shell vials.
- Specimen: Nasopharyngeal swab or aspirate in viral transport media (e.g., M4).
- Growth Media: Cell line-specific maintenance media (e.g., MEM, DMEM) supplemented with antibiotics (Penicillin/Streptomycin) and antifungals (Amphotericin B).
- Trypsin: For MRC-5 cells used in influenza isolation, add TPCK-trypsin to a final concentration of 1-2 µg/mL [32].
- Fixation and Staining Reagents: Acetone, virus-specific monoclonal antibodies, and fluorescent-labeled secondary antibodies.
Procedure:
- Specimen Preparation: Vortex the clinical specimen in transport media and clarify by low-speed centrifugation (~500 x g for 10 minutes).
- Inoculation: Aseptically remove growth media from the cell cultures and inoculate each cell line with 0.2-0.5 mL of the supernatant from the prepared specimen. Include negative control cultures inoculated with maintenance media only.
- Adsorption: Incubate the inoculated cultures at 33-35°C for 60-90 minutes to allow for viral adsorption. Rock the cultures every 15-20 minutes.
- Maintenance: After adsorption, add maintenance media to the cultures. For MRC-5 cells used for influenza, ensure the maintenance media contains trypsin [32]. Incubate all cultures at 33-35°C and observe daily for Cytopathic Effect (CPE).
- Observation and Monitoring:
- RhMK cells: Examine daily for RSV-specific CPE (e.g., syncytia formation). According to studies, 50% of positive RSV cultures show CPE by day 5 and 90% by day 7 [30].
- MRC-5 and HEp-2 cells: Monitor for cell rounding and degeneration. CPE development in MRC-5 cells may be slower than in RhMK for RSV [30].
- Confirmation: Upon observation of CPE or at a predetermined endpoint (e.g., 7-10 days post-inoculation), confirm the presence of virus by immunofluorescence assay (IFA) or PCR. For IFA, scrape the cell monolayer, spot onto slides, fix with acetone, and stain with virus-specific antibodies.
Protocol 2: Optimized Isolation of Adenovirus and Herpes Simplex Virus using A549 Cells
The A549 cell line demonstrates high susceptibility to adenovirus and HSV, making it a superior choice for isolating these pathogens [33].
Application: Isolation of Adenovirus and Herpes Simplex Virus (HSV). Key Cell Lines: A549.
Materials and Reagents:
- Cell Line: A549 cells at a low passage number (passage <120 recommended to maintain optimal susceptibility) [33].
- Specimen: Throat swab (for adenovirus) or lesion swab (for HSV) in viral transport media.
- Growth Media: DMEM or EMEM supplemented with fetal bovine serum (FBS, 2-10%), L-glutamine, and antibiotics.
Procedure:
- Cell Preparation: Use A549 cells that are 80-100% confluent. Avoid using cells at high passage numbers (>120) as sensitivity to adenovirus may decline [33].
- Inoculation: Follow the specimen preparation and adsorption steps as described in Protocol 1.
- Incubation and Monitoring: Incubate inoculated A549 cultures at 36°C and observe daily for CPE.
- Confirmation: Confirm the viral identity by IFA, as described in Protocol 1. The high susceptibility of A549 cells often leads to rapid and extensive CPE, facilitating easy detection.
Visualizing the Virus Isolation Workflow
The following diagram illustrates the logical decision-making process for selecting the appropriate cell line based on the suspected viral pathogen, as derived from the protocols and data above.
Diagram Title: Cell Line Selection for Virus Isolation
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues the essential materials and reagents required to establish a robust viral culture system using the featured cell lines.
Table 2: Essential Reagents for Virus Isolation in Cell Culture
| Reagent/Cell Line | Function / Application | Specific Notes and Considerations |
|---|---|---|
| Primary RhMK Cells | Isolation of RSV, influenza, and parainfluenza viruses. | High susceptibility to RSV; CPE develops rapidly. Limited lifespan and potential for endogenous viral contaminants [30]. |
| MRC-5 Cell Strain | Isolation of influenza, RSV, and cytomegalovirus. | Human diploid fibroblast; reliable and standardized. Requires trypsin supplementation in media for optimal influenza isolation [30] [32]. |
| HEp-2 Cell Line | Benchmark cell line for isolation of Human RSV (HRSV). | Provides consistent results for HRSV. Often used in combination with other cell lines to maximize detection sensitivity [34]. |
| A549 Cell Line | Highly sensitive isolation of adenovirus and HSV. | Efficient and economical alternative. Monitor passage number; sensitivity may decrease after passage 120 [33]. |
| Viral Transport Media (e.g., M4) | Preserves viral viability during specimen transport. | Essential for maintaining sample integrity from collection to laboratory inoculation [31]. |
| TPCK-Trypsin | Cleaves influenza hemagglutinin, enabling multi-cycle replication. | Critical supplement for culturing influenza virus in MRC-5 and other non-enterocytic cells [32]. |
| Virus-Specific Monoclonal Antibodies | Confirmation and identification of isolated viruses via IFA. | Allows for rapid, specific typing of the virus causing CPE in the culture [34]. |
Practical Protocols: Traditional and Advanced Techniques for Virus Cultivation
Virus isolation in cell culture remains a foundational technique in clinical virology and viral research, providing a means to detect, amplify, and identify infectious viral pathogens. Within this domain, three methodological approaches have evolved to address differing needs for throughput, speed, and scalability: conventional tube cultures, shell vial cultures, and microtiter plate-based techniques. These methods serve as critical tools for diagnosing infections, conducting epidemiological studies, and supporting drug and vaccine development. Despite the emergence of molecular detection methods, virus isolation retains irreplaceable value for confirming active infection, obtaining viral isolates for characterization, and evaluating antiviral efficacy. This application note details the protocols, applications, and performance characteristics of these three standard isolation methods within the broader context of cell culture methodologies for virus research.
Methodological Principles and Comparative Analysis
Technical Foundations
Conventional Tube Cultures (TC): This traditional method involves inoculating clinical specimens onto cell monolayers in culture tubes, which are then incubated for days to weeks and monitored periodically for cytopathic effect (CPE). It is considered a "gold standard" for its ability to detect a wide spectrum of viruses but is limited by long turnaround times [36] [37].
Shell Vial Cultures (SV): Developed to accelerate viral detection, this centrifugation-enhanced assay uses small vials containing a coverslip with a cell monolayer. Specimens are centrifuged onto the monolayer to enhance viral adsorption, followed by incubation for 16-48 hours and subsequent immunostaining for early viral antigens. This method significantly reduces detection time compared to conventional tube cultures [36] [37] [38].
Microtiter Plate-Based Isolation: This high-throughput approach adapts virus isolation to 96-well plate formats, allowing parallel processing of numerous samples. After incubation, viral presence is typically detected using immunostaining assays such as immunoperoxidase monolayer assay (IPMA) or monolayer enzyme-linked immunosorbent assay (M-ELISA) [39] [40].
Performance Comparison of Isolation Methods
The following table summarizes the performance characteristics of these methods for detecting various viruses across published studies:
Table 1: Comparative Performance of Virus Isolation Methods
| Virus Detected | Method | Detection Time | Sensitivity (%) | Specificity (%) | Key Findings |
|---|---|---|---|---|---|
| Cytomegalovirus (CMV) | Shell Vial (MRC-5 cells) | 16 hours | 100 | 100 | Detected 124 positives vs. 88 by TC; more sensitive than TC (9-day average) [36] |
| Respiratory Syncytial Virus (RSV) | Shell Vial (CoHLM cells) | 48 hours | 94.1 (160/170 strains) | N/R | Detected 160 of 170 strains vs. 167 by TC (mean 6 days) [37] |
| Various Respiratory Viruses* | Shell Vial | 48 hours | 95.2 | N/R | Overall detection of 160/170 isolates; TC detected 167/170 [37] |
| Influenza A & B | Shell Vial | 48 hours | 100 (18/18, 4/5) | N/R | Detected all 18 Flu A and 4 of 5 Flu B isolates [38] |
| Adenovirus | Shell Vial | 48 hours | 47.6 (10/21) | N/R | Shell vials were ineffective for adenovirus compared to TC [38] |
| Bovine Viral Diarrhea Virus (BVDV) | Microtiter IPMA/M-ELISA | 4 days | 85 (100 for PI^) | 100 | Relative to standard VI; required only 4 days of incubation [39] |
*Respiratory viruses include RSV, influenza A and B, parainfluenza 1-3, and adenovirus. ^PI: Persistently infected cattle.
Workflow and Decision Pathway
The following diagram illustrates the procedural workflows for the three virus isolation methods and a decision pathway for selecting the appropriate technique:
Detailed Experimental Protocols
Protocol 1: Shell Vial Culture for Respiratory Viruses
Principle: Centrifugation-enhanced infection of mixed cell monolayers on coverslips enables rapid detection of multiple respiratory viruses through immunofluorescence staining within 48 hours [37].
Materials:
- Shell vials (1-dram, ~3.7 ml) with 12-mm coverslips
- Cell lines: HEp-2, LLC-MK2, and MDCK
- Modified Eagle Minimum Essential Medium (MEM) with 10% fetal bovine serum (FBS)
- Clinical specimens (nasal wash, nasopharyngeal aspirates)
- Virus-specific monoclonal antibodies (e.g., Respiratory Viral Screen IFA)
- Fluorescence microscope
Procedure:
- Cell Monolayer Preparation:
- Prepare separate suspensions of HEp-2, LLC-MK2, and MDCK cells at 150,000 cells/ml.
- Create a mixed cell suspension containing 50,000 cells/ml of each cell type.
- Add 1 ml of the mixed cell suspension to each shell vial containing a coverslip.
- Incubate vials at 37°C for 24 hours to form confluent monolayers.
Specimen Processing:
- Sonicate nasal wash specimens for 1 minute.
- Centrifuge at 500 × g for 5 minutes to obtain cell-free supernatant.
Inoculation and Centrifugation:
- Inoculate 0.2 ml of specimen supernatant onto each of two shell vials.
- Centrifuge vials at 3,500 × g for 15 minutes at 25°C.
- Incubate at 37°C for 1 hour for adsorption.
Maintenance and Incubation:
- Discard supernatant and add 1 ml of maintenance MEM with 1% FBS and 0.2 μg/ml trypsin.
- Incubate vials at 37°C with continuous shaking for 48 hours.
Virus Detection and Identification:
- After 48 hours, remove one vial and fix the coverslip monolayer with cold (-20°C) acetone.
- Stain with pooled monoclonal antibodies against respiratory viruses (RSV, adenovirus, influenza A/B, parainfluenza 1-3).
- If fluorescent cells are observed, fix and stain the second vial with virus-specific monoclonal antibodies for final identification.
- Examine under fluorescence microscope at ×250 magnification.
Protocol 2: Microtiter Plate Virus Isolation with Immunostaining
Principle: Cell culture in 96-well microtiter plates enables high-throughput virus isolation with detection via immunoperoxidase or ELISA-based methods, ideal for screening large sample numbers [39].
Materials:
- 96-well flat-bottom microtiter plates
- Appropriate cell line (e.g., bovine cells for BVDV)
- Growth and maintenance media
- Serum samples for testing
- Virus-specific monoclonal antibodies
- Species-specific peroxidase conjugate
- Enzyme substrate (e.g., AEC for IPMA, ABTS for M-ELISA)
- Plate reader (for M-ELISA)
Procedure:
- Cell Seeding:
- Prepare cell suspension at appropriate concentration (e.g., 1×10^5 cells/ml).
- Dispense 100 μl/well into 96-well microtiter plates.
- Incubate at 37°C with 5% CO₂ until confluent monolayers form (24-48 hours).
Sample Inoculation:
- Remove growth medium from wells.
- Inoculate test serum samples (10-20 μl/well) in duplicate or triplicate.
- Include appropriate positive and negative controls.
- Incubate plates at 37°C for 1 hour for adsorption.
Maintenance and Incubation:
- Add maintenance medium to wells.
- Incubate plates at 37°C with 5% CO₂ for 4 days.
Immunoperoxidase Monolayer Assay (IPMA):
- Remove medium and wash cells with phosphate-buffered saline (PBS).
- Fix cells with 20-30% acetone for 10 minutes.
- Add virus-specific monoclonal antibody and incubate for 30-60 minutes.
- Wash and add species-specific peroxidase conjugate for 30-60 minutes.
- Add enzyme substrate (AEC) and incubate until color develops.
- Examine for red intracellular precipitate under microscope.
Monolayer ELISA (M-ELISA) Alternative:
- After fixation, follow similar antibody binding steps as IPMA.
- Use substrate that produces soluble colored product (e.g., ABTS).
- Measure optical density with plate reader for objective results.
Protocol 3: Conventional Tube Culture for Virus Isolation
Principle: Inoculation of specimens onto cell monolayers in culture tubes with extended incubation allows detection of a wide range of viruses through observation of cytopathic effects, serving as a reference standard [36] [41].
Materials:
- Cell culture tubes with appropriate cell lines (e.g., MRC-5, WI38, rhesus monkey kidney)
- Maintenance medium (Eagle's minimum essential medium)
- Clinical specimens (urine, blood, tissue, respiratory secretions)
- Inverted microscope
Procedure:
- Cell Monolayer Preparation:
- Select appropriate cell lines based on target viruses.
- Ensure confluent, healthy monolayers in culture tubes before inoculation.
Specimen Inoculation:
- Inoculate 4 drops of antibiotic-treated specimen onto cell monolayer.
- Incubate at 37°C for 1 hour to allow viral adsorption.
Maintenance and Observation:
- Add 1.5 ml of maintenance medium to each tube.
- Incubate tubes at 37°C and examine daily for cytopathic effect (CPE) using an inverted microscope.
- Continue observation for up to 14 days, depending on suspected virus.
Hemadsorption (for certain viruses):
- For tubes without CPE at days 5-10, perform hemadsorption with guinea pig erythrocytes.
- Scrape cells and test with specific immunofluorescence if hemadsorption positive.
Virus Identification:
- Once CPE is observed, identify virus using type-specific immunofluorescence.
- Subculture to fresh tubes for virus propagation if needed.
Research Reagent Solutions
Table 2: Essential Research Reagents for Virus Isolation Methods
| Reagent/Cell Line | Application | Function/Purpose | Example Use Cases |
|---|---|---|---|
| MRC-5 Cells | Virus Isolation | Human diploid lung fibroblast; sensitive to many human viruses | CMV detection [36], general viral diagnosis |
| HEp-2 Cells | Respiratory Virus Isolation | Human laryngeal carcinoma; sensitive to RSV and adenoviruses | Component of CoHLM mixed cell system [37] |
| LLC-MK2 Cells | Respiratory Virus Isolation | Rhesus monkey kidney; sensitive to influenza and parainfluenza | Component of CoHLM mixed cell system [37] |
| MDCK Cells | Influenza Isolation | Canine kidney; optimal for influenza A and B propagation | Component of CoHLM mixed cell system [37] |
| Virus-Specific Monoclonal Antibodies | Viral Detection & Identification | Immunological recognition of specific viral antigens | Early antigen detection in shell vials [36] |
| Pooled Monoclonal Antibodies | Viral Screening | Simultaneous detection of multiple respiratory viruses | Respiratory Viral Screen IFA [37] |
| Fluorescein-Labelled Conjugates | Immunofluorescence | Fluorescent detection of antibody-bound viral antigens | Shell vial staining [37] [38] |
| Peroxidase Conjugates | Immunostaining | Enzymatic detection for colorimetric visualization | IPMA and M-ELISA [39] |
Standard virus isolation methods including tube cultures, shell vial assays, and microtiter plate techniques provide a hierarchy of options balancing throughput, speed, and detection breadth. Conventional tube culture remains the comprehensive reference method despite its extended timeline. Shell vial cultures with centrifugation-enhancement offer an optimal balance of speed (24-48 hours) and sensitivity for many clinical applications, particularly for cytomegalovirus and respiratory viruses like RSV and influenza. Microtiter plate-based systems excel in high-throughput scenarios requiring standardized processing of large sample numbers. Method selection depends on specific application requirements: tube cultures for broad detection where time is secondary, shell vials for rapid clinical diagnosis, and microtiter methods for large-scale screening programs. Together, these established isolation methods continue to provide indispensable tools for both clinical virology and pharmaceutical research, maintaining relevance alongside molecular techniques by delivering biologically active virus isolates essential for pathogenesis studies, antiviral development, and vaccine production.
Optimizing Sample Collection, Processing, and Inoculation Techniques
Within the framework of virus isolation research, the pathway to successful cell culture begins long before a specimen enters the biosafety cabinet. The pre-analytical phase—encompassing sample collection, processing, and the preparation of inoculum—is a critical determinant of experimental success. Errors introduced during these initial steps can lead to false negatives, compromised cell cultures, or a complete failure to isolate the viable virus, thereby invalidating subsequent research efforts. This document provides detailed application notes and protocols designed to standardize and optimize these foundational techniques. By implementing these evidence-based procedures, researchers can enhance the sensitivity, reliability, and reproducibility of their viral isolation studies, ensuring that high-quality data flows from robust methodological beginnings.
Optimizing Sample Collection and Transport
The integrity of viral isolation research is fundamentally dependent on the initial collection and stabilization of specimens. The choice of tools, media, and handling conditions directly influences the recovery of viable viral particles.
Selection of Sample Collection Devices
The physical properties of the collection swab significantly impact the release of viral material into transport media. The following table summarizes key findings from a comparative study on sample collection devices:
Table 1: Comparison of Sample Collection Device Efficacy for Virus Recovery
| Device Type | Viral RNA Detection Rate (%) | Geometric Mean Titer (Log10 EID50 Equivalents per 25 cm²) | Key Characteristics |
|---|---|---|---|
| Pre-moistened Cotton Gauze | 100% | 3.2 | Superior sample absorption and elution; optimal for environmental surface sampling [42]. |
| Foam Swab | 95% | 2.8 | Effective recovery; often used in clinical and veterinary settings [42] [43]. |
| Flocked Nylon Swab | Not Specified | Not Specified | Consistently performs well with transport media; superior sample release due to perpendicular fibers [43]. |
| Dry Cotton Gauze | 93% | 2.6 | Lower recovery and detection rates compared to pre-moistened and foam alternatives [42]. |
| Non-flocked Dacron Swab | Not Specified | Not Specified | Inferior recovery compared to flocked and foam swabs [43]. |
Choice of Transport Media and Conditions
The chemical composition and volume of the transport medium are crucial for preserving viral viability during transit.
- Media Formulation: Brain Heart Infusion (BHI) broth has been demonstrated to be superior to Phosphate-Buffered Saline (PBS) for maintaining virus stability, leading to a higher number of positive virus isolations [43]. The medium must contain antibiotics and protein (e.g., albumin or serum) to prevent microbial overgrowth and stabilize viral particles.
- Media Volume: While 2-3.5 mL is common, a larger volume (e.g., 3.5 mL) may improve detection sensitivity for samples with low viral load, as it demonstrates a trend towards more positive results at later time points post-infection [43].
- Wet vs. Dry Transport: Transporting swabs in media is consistently better for virus recovery and detection than transporting them dry. Leaving the swab in the media vial during transport is recommended, as removing it does not improve recovery and may marginally decrease it [43].
Timing and Specimen Handling
- Timing of Collection: Specimens should be collected as early as possible following the onset of clinical signs, ideally within the first week, when viral shedding is typically highest [11].
- Temperature Control: Specimens must be refrigerated immediately after collection and transported to the laboratory on wet ice or cold packs as quickly as possible, preferably within 24 hours [11]. For delays exceeding 2-3 days, freezing at -70°C is preferable to -20°C to minimize loss of viability [11]. Note that for molecular detection from wastewater, storage at 4°C has been shown to enhance SARS-CoV-2 detection compared to -20°C [44].
Figure 1: Optimized Workflow for Viral Sample Collection and Transport
Sample Processing and Nucleic Acid Isolation
Efficient release and purification of nucleic acids are prerequisites for sensitive molecular detection and characterization. The following protocol, adapted for a variety of sample types, ensures high-quality extracts.
CTAB-Based Protocol for Complex Samples
This method is particularly effective for complex or difficult samples, such as plant tissues stored in silica gel, and produces nucleic acids of high quality suitable for PCR, RT-PCR, and sequencing [45].
Table 2: Key Reagents for Nucleic Acid Isolation via CTAB Protocol
| Reagent | Function | Specifications/Alternatives |
|---|---|---|
| CTAB (Cetyltrimethylammonium bromide) | Lysis buffer; complexes with polysaccharides to remove them during purification. | Use a concentration of 2% (w/v) in the extraction buffer [45]. |
| PVP (Polyvinylpyrrolidone) | Binds polyphenols, preventing co-purification and inhibition of downstream enzymes. | Use a concentration of 2% (w/v); molecular weight PVP-10 is typical [45]. |
| 2-Mercaptoethanol (βME) | Reducing agent; helps to denature proteins and inhibit oxidation of polyphenols. | Add to CTAB buffer just before use (e.g., 200 µL per 100 mL) in a fume hood [45]. |
| Chloroform | Organic solvent for liquid-phase separation; denatures and removes proteins. | Use ice-cold; always handle in a fume hood [45]. |
| Isopropanol | Precipitates nucleic acids from the aqueous phase. | Use ice-cold for higher yield [45]. |
| Silica Gel | Desiccant for rapid drying and preservation of tissue samples prior to extraction. | Preserves nucleic acid integrity during storage and transport [45]. |
Detailed Protocol:
- Homogenization: Transfer 20 mg of dried tissue to a 2 mL microfuge tube containing a carbon steel ball and grind for 3 minutes using a paint shaker or similar mechanical homogenizer at room temperature [45].
- Initial Lysis: Add 1 mL of pre-warmed (65°C) CTAB extraction buffer (containing 2% βME) to the powdered tissue. Vortex vigorously until a homogeneous suspension forms. Centrifuge at 13,000 RPM for 3 minutes at room temperature [45].
- Secondary Lysis and Debris Removal: Transfer 750 µL of the supernatant to a new 2 mL tube. Incubate at 65°C for 15 minutes, then place on ice [45].
- Chloroform Cleanup: Add 750 µL of ice-cold chloroform to the lysate. Vortex for 1 minute to form an emulsion. Centrifuge at 13,000 RPM for 10 minutes at 4°C [45].
- Nucleic Acid Precipitation: Transfer the upper aqueous phase to a new 1.5 mL tube. Add an equal volume (approx. 750 µL) of ice-cold isopropanol. Mix by gentle inversion and incubate at -20°C for at least 30 minutes [45].
- Pellet Washing: Centrifuge at 13,000 RPM for 15 minutes at 4°C to pellet the nucleic acids. Discard the supernatant. Wash the pellet with 500 µL of 70% ice-cold ethanol and centrifuge again at 13,000 RPM for 5 minutes. Discard the ethanol and air-dry the pellet for 15 minutes [45].
- Resuspension: Resuspend the pellet in 50 µL of nuclease-free water. Assess the quality and concentration using a spectrophotometer (e.g., Nanodrop). Absorbance ratios of OD260/280 above 1.8 and OD260/230 above 1.7 indicate a pure preparation [45].
Alternative Methods and Considerations
- Magnetic Bead-Based Kits: For high-throughput applications, kits like the MagMAX-96 viral RNA isolation kit have shown excellent recovery rates (e.g., 50.1 ± 20.1% for hCoV-229E) and are amenable to automation [44].
- pH Adjustment for Environmental Samples: For wastewater and other challenging environmental matrices, pre-adjusting the sample pH to 9.6 can aid in virus desorption from solids, followed by a return to neutral pH after solid removal, which significantly increases viral recovery [44].
- DNase/RNase Treatment: For RNA virus detection, treat the total nucleic acid extract with DNase I. For DNA virus detection, RNase A treatment is generally unnecessary prior to PCR [45].
Preparation of Inoculum and Mechanical Inoculation
The transition from a raw sample to a prepared inoculum is a critical step for successful virus isolation in cell culture or other host systems.
Protocol for Mechanical Inoculation in Plant Virus Research
This optimized protocol for Watermelon Bud Necrosis Virus (WBNV) demonstrates principles applicable to other hard-to-transmit viruses [46].
Detailed Protocol:
- Inoculum Source Preparation: Use young, symptomatic leaf tissue stored at -80°C as the virus source [46].
- Extraction Buffer: Grind tissue in 0.1 M Potassium Phosphate Buffer (pH 7.0), containing 0.2% Sodium sulfite and 0.01 M 2-Mercaptoethanol, using a pre-chilled mortar and pestle [46].
- Abrasive Application: Prior to inoculation, dust the carborundum (600-mesh) evenly onto the surface of the recipient plant's leaves (e.g., at the 2-4 leaf stage for watermelon) to create micro-wounds [46].
- Inoculation Technique: Dip a sterile cotton swab or forefinger into the prepared inoculum and gently rub it onto the dusted leaves. Apply consistent, mild pressure in a unidirectional motion [46].
- Post-Inoculation Rinsing: Approximately 5 minutes post-inoculation, gently rinse the leaves with distilled water to remove excess abrasive and inoculum, minimizing plant tissue damage [46].
Processing Clinical Specimens for Cell Culture Inoculation
For clinical samples (e.g., swab media, tissue homogenates, fecal samples), the following steps are essential:
- Clarification: Centrifuge the sample at low speed (e.g., 2,000 - 5,000 × g for 10 minutes) to remove coarse debris and bacterial cells. This step is critical to prevent contamination of the cell culture.
- Filtration: Filter-sterilize the supernatant through a 0.22 µm or 0.45 µm pore-size membrane. This removes bacteria and fungi, but allows most viral particles to pass through.
- Antibiotic Addition: Supplement the inoculum with antibiotics (e.g., Penicillin-Streptomycin, Amphotericin B) to further control microbial contamination.
Figure 2: Sample Processing and Inoculum Preparation Workflow
Biosafety and Quality Control
Maintaining biosafety and quality control throughout these procedures is non-negotiable.
- Biosafety Level (BSL): At a minimum, BSL-2 facilities, practices, and procedures are recommended for diagnostic research and virus propagation with human pathogens like SARS-CoV-2. A comprehensive, activity-specific risk assessment must be performed [47].
- Aerosol-Generating Procedures: Procedures with a high likelihood of generating aerosols or droplets (e.g., pipetting, centrifuging, vortexing, sonicating) should be conducted within a certified Class II Biological Safety Cabinet (BSC) [47].
- Decontamination: Work surfaces and equipment must be decontaminated with appropriate disinfectants. Use EPA-registered disinfectants qualified for use against the specific virus being handled, following the manufacturer's recommended contact time [47].
- Inhibition Monitoring: Include internal controls in molecular assays. For example, using salmon DNA to monitor PCR inhibition or an exogenous control virus to track extraction efficiency ensures result reliability [44].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagent Solutions for Viral Sample Processing and Isolation
| Reagent / Kit | Primary Function | Application Notes |
|---|---|---|
| CTAB Extraction Buffer | Total nucleic acid isolation from complex samples (tissues, plants). | Effective for removing polysaccharides and polyphenols; ideal for difficult samples stored in silica gel [45]. |
| MagMAX-96 Viral RNA Isolation Kit | High-throughput, automated RNA purification. | Showed superior recovery (50.1%) of human coronavirus 229E; ideal for liquid samples like wastewater [44]. |
| Flocked Nylon Swabs | Clinical and environmental sample collection. | Superior sample release into transport media compared to traditional fiber-wound swabs [43]. |
| Brain Heart Infusion (BHI) Broth | Viral transport medium. | Demonstrated to be superior to PBS for maintaining viability of avian influenza and Newcastle disease virus [43]. |
| Potassium Phosphate Buffer (with additives) | Extraction buffer for mechanical inoculation of plant viruses. | Buffer containing sodium sulfite and 2-mercaptoethanol is critical for successful transmission of labile viruses like WBNV [46]. |
| One-step RT-qPCR Master Mix | Direct detection and quantification of viral RNA. | Offers improved sensitivity for SARS-CoV-2 detection compared to two-step methods and simplifies the workflow [44]. |
| DNase I (RNase-free) | Removal of genomic DNA contamination from RNA extracts. | Essential step for accurate RNA virus detection via RT-PCR when using total nucleic acid extraction protocols [45]. |
The evolution of cell culture methodologies represents a critical frontier in virology and drug development research. Traditional two-dimensional (2D) monolayer cultures, while useful for high-throughput screening, often fail to recapitulate the complex physiological environment required for accurate viral studies [48]. In response, the field has advanced significantly towards more sophisticated systems. This document details two such innovative approaches: the use of cryopreserved cell cultures for ensuring experimental reproducibility and flexibility, and the development of complex co-culture systems that model the multicellular interactions of host tissues. These methodologies are particularly transformative for virus isolation research, enabling more precise studies of viral pathogenesis, host-pathogen interactions, and therapeutic efficacy [1].
Cryopreserved Cell Cultures in Virology
Cryopreservation allows for the long-term storage of viable cells by cooling them to sub-zero temperatures, typically in liquid nitrogen at -196°C [1]. This technique has moved beyond simple cell banking to become a fundamental tool in virology. Its primary advantages include the standardization of cell batches, which reduces inter-experiment variability, and the creation of ready-to-use cellular resources for rapid response during viral outbreaks. Furthermore, it facilitates the sharing of rare or precious cell lines between laboratories, enhancing collaborative research efforts [49].
Quantitative Impact on Cell Viability and Function
The success of cryopreservation is contingent on optimized protocols. Deviations can significantly impact cellular viability and function, as summarized in the table below.
Table 1: Impact of Cryopreservation Parameters on PBMC Viability and Function
| Parameter | Optimal Condition (HANC-SOP) | Suboptimal Condition | Observed Effect on PBMCs | Citation |
|---|---|---|---|---|
| Processing Time | ≤ 8 hours | 24 hours or more | Reduced cell viability | [49] |
| Storage Temperature | -196°C (Liquid N₂) | Fluctuating temperatures | Reduced viability and immunogenicity | [49] |
| Cryomedium | 10% DMSO | N/A | Preserved Treg immunosuppressive function | [50] |
| Post-Thaw Resting | 24 hours | Immediate stimulation | Restored natural immunogenic capabilities | [49] |
Detailed Protocol: Cryopreservation and Thawing of PBMCs
This protocol is adapted from the HANC Cross-Network PBMC Processing SOP and the IMPAACT PBMC Thawing SOP, which are considered gold standards in the field [49].
Cryopreservation
- Materials:
- Separation Medium: Ficoll-Paque or Lymphoprep.
- Cryopreservation Medium: Roswell Park Memorial Institute (RPMI) medium or phosphate-buffered saline (PBS) supplemented with 10% Dimethyl Sulfoxide (DMSO) and 10-20% Fetal Bovine Serum (FBS). For clinical settings, human serum albumin is preferred [50] [49].
- Procedure:
- Isolate PBMCs from peripheral blood using density-gradient centrifugation with SepMate tubes or similar. Centrifuge at 400 × g for 10 min, then layer cell suspension over separation medium and centrifuge at 1200 × g for 10 min with brake [50] [49].
- Wash and Count the collected PBMC fraction with PBS. Centrifuge at 350-400 × g for 10 min. Assess cell concentration and viability using a NucleoCounter or Trypan Blue exclusion [50].
- Resuspend the cell pellet in cryopreservation medium at a concentration of < 50 × 10⁶ cells/mL [50].
- Aliquot 1 mL of cell suspension into cryovials and place them in a controlled-rate freezing container (e.g., Corning CoolCell) [50].
- Transfer vials to a -80°C freezer for >24 hours, then to long-term storage in liquid nitrogen [50] [49].
Thawing and Post-Thaw Recovery
- Materials: Water bath (37°C), RPMI medium with 5% FBS.
- Procedure:
- Rapid Thaw: Remove vial from liquid nitrogen and partially thaw in a 37°C water bath for approximately 1 minute [50] [49].
- Gentle Dilution: Transfer cell suspension to a 15 mL tube and gradually add 12 mL of pre-warmed RPMI medium with 5% FBS at spaced intervals [50].
- Wash: Centrifuge at 300-400 × g for 10 min. Discard supernatant and repeat wash step [50] [49].
- Rest Cells: Resuspend the final pellet in complete medium and incubate at high density (e.g., 1-2 × 10⁶ cells/mL) for 24 hours before experimental use. This resting period is critical for recovering surface receptor expression and normal cellular function [49].
Diagram 1: PBMC cryopreservation and thawing workflow.
Co-cultured Systems for Advanced Viral Research
Co-culture systems involve growing two or more different cell types together to create a more physiologically relevant model than is possible with monocultures. These systems are pivotal for investigating the complex dynamics of the tumor microenvironment (TME) and virus-host interactions [51]. By incorporating immune cells, fibroblasts, or other stromal components, co-cultures can replicate critical biological processes such as immune cell recruitment, cytokine signaling, and the breakdown of epithelial barriers during infection [48] [51].
Key Applications in Viral Co-infections and Immunobiology
Advanced co-culture models have yielded significant insights, as shown in the following table.
Table 2: Applications of Co-culture Systems in Viral and Immunological Research
| Co-culture Model | Research Application / Finding | Key Outcome / Insight | Citation |
|---|---|---|---|
| A549-hACE2 + Viral Co-infection | Study of RSV and SARS-CoV-2 co-infection dynamics | RSV replication increased due to upregulated ICAM1 receptor, pro-inflammatory signaling, and disrupted autophagy. | [48] |
| Tumor Organoids + PBMCs | Enrichment of tumor-reactive T cells from patient blood. | Effective method to assess cytotoxic T cell efficacy against matched tumor organoids on an individual patient level. | [51] |
| Pancreatic Cancer Organoids + PBMCs | Modeling tumor-immune interactions. | Observed activation of cancer-associated fibroblasts and tumor-dependent lymphocyte infiltration. | [51] |
| R-Mix (A549 + Mink Lung Cells) | Multiplexed detection of respiratory viral pathogens. | Sensitive and rapid identification of viruses like influenza, RSV, and adenoviruses within 24 hours. | [1] |
Detailed Protocol: Establishing a Tumor Organoid-Immune Cell Co-culture
This protocol simulates the tumor microenvironment to study immune cell infiltration and function [51].
- Materials:
- Extracellular Matrix (ECM): Matrigel or other biocompatible scaffolds.
- Organoid Culture Medium: Advanced media (e.g., DMEM/F12) supplemented with specific growth factors (e.g., Wnt3A, R-spondin-1, Noggin, EGF).
- Immune Cells: Peripheral blood lymphocytes (PBLs) or PBMCs isolated from patient blood or buffy coats.
- Procedure:
- Establish Tumor Organoids: a. Mechanically dissociate and enzymatically digest patient tumor samples. b. Seed the cell suspension onto a pre-polymerized Matrigel bed. c. Culture in organoid-specific medium to promote 3D structure formation and maintenance [51].
- Isolate Immune Cells: a. Isolate PBMCs from donor blood using the density-gradient centrifugation method described in Section 2.3.1. b. If needed, further isolate specific immune cell populations (e.g., T cells) using magnetic-activated cell sorting (MACS) with negative selection (LD column) and positive selection (e.g., CD4+ CD25+ beads for Tregs) [50].
- Initiate Co-culture: a. Once organoids are established (typically after 5-7 days), harvest and partially dissociate them into smaller clusters. b. Seed the organoid clusters into a new Matrigel droplet or a low-attachment plate. c. Add the isolated immune cells directly to the culture medium at the desired effector-to-target ratio (e.g., 1:1, 1:0.5) [51].
- Maintenance and Analysis: a. Culture the co-culture system for the desired duration (e.g., 5 days), refreshing medium as needed. b. Monitor immune cell infiltration and organoid viability using live-cell imaging, immunohistochemistry, or flow cytometry. Functional readouts can include cytokine release assays and assessment of organoid death [51].
Diagram 2: Tumor organoid-immune cell co-culture setup.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues critical reagents and their functions for executing the protocols described in this document.
Table 3: Essential Reagents for Cryopreservation and Co-culture Workflows
| Reagent / Material | Function / Application | Specific Example / Note | |
|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Cryoprotectant that prevents intracellular ice crystal formation. | Used at 10% final concentration in cryopreservation medium. | [50] [49] |
| Ficoll-Paque / Lymphoprep | Density gradient medium for isolating PBMCs from whole blood. | Enables separation of mononuclear cells from granulocytes and red blood cells. | [50] [49] |
| Matrigel | Basement membrane extract providing a 3D scaffold for organoid growth. | Essential for supporting the structural and functional polarity of organoids. | [51] |
| Roswell Park Memorial Institute (RPMI) 1640 Medium | Standard cell culture medium for lymphocytes and many other cell types. | Commonly used for culturing PBMCs and in co-culture experiments. | [50] [49] |
| Growth Factor Cocktails | Supplements for specialized media to maintain stemness and drive differentiation. | For organoids: Wnt3A, R-spondin-1, Noggin, Epidermal Growth Factor (EGF). | [51] |
| Magnetic Cell Separation Kits (MACS) | Isolation of highly pure specific cell populations (e.g., T cells). | Kits typically include biotinylated antibodies and microbeads for positive/negative selection. | [50] |
| CellTrace Violet Proliferation Kit | Fluorescent dye to track and quantify cell division over time. | Used in functional assays to measure T cell proliferation in response to stimulation. | [50] |
Specialized Methods for Cell-Associated Virus Isolation and Particle Release
Within the broader context of cell culture methods for virus isolation research, the study of cell-associated viruses presents unique challenges. Unlike viruses that are freely released into the surrounding medium, cell-associated viruses, such as the MX strain of lymphocytic choriomeningitis virus (LCMV), are preferentially propagated by cell-to-cell contact and do not release distinct virions in large quantities [52]. This characteristic necessitates specialized isolation techniques to effectively release and study infectious viral particles from infected host cells. Traditional methods involving a combination of hypotonic burst, freeze-thaw cycles, and sonication are often employed but may not always yield optimal results. This application note provides a detailed comparison of established and optimized protocols for the isolation of cell-associated viral particles, framed within the critical need for robust and efficient methodologies in virology research and drug development.
Comparative Analysis of Virus Isolation Methods
The selection of an appropriate virus isolation method is critical for downstream applications, including viral quantification, pathogenicity studies, and vaccine development. The following section provides a structured comparison of different techniques, highlighting their performance, advantages, and limitations.
Table 1: Comparison of Virus Isolation and Detection Methods
| Method Category | Specific Technique | Target Virus/Application | Reported Performance/Sensitivity | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Cell-Associated Particle Release | Deionized Water Lysis [52] | Lymphocytic Choriomeningitis Virus (LCMV) MX strain | Most effective method for releasing infectious particles [52] | Fast, simple, avoids potential damage from freeze-thaw/sonication [52] | Primarily demonstrated for LCMV MX; efficacy for other viruses requires validation. |
| Cell-Associated Particle Release | Freeze-Thaw Cycles & Sonication [52] | Lymphocytic Choriomeningitis Virus (LCMV) MX strain | Does not improve virus isolation compared to water lysis alone [52] | Common traditional approach. | Can be time-consuming; may not enhance yield. |
| Viral RNA Isolation from Complex Samples | Zymo Environ Water RNA Kit [53] | SARS-CoV-2 in Wastewater | Superior RNA quality compared to PEG precipitation and ultrafiltration [53] | Combines viral enrichment and RNA purification; works well without sample filtration [53] | Optimized for environmental water; may require adaptation for cell culture lysates. |
| Viral RNA Isolation from Complex Samples | PEG Precipitation + NucleoSpin RNA Virus Kit [53] | SARS-CoV-2 in Wastewater | Lower RNA quality compared to direct isolation kit [53] | Common and accessible protocol for virus concentration. | Requires overnight incubation; more processing steps. |
| Viral RNA Isolation from Complex Samples | Ultrafiltration (Vivaspin) + NucleoSpin RNA Virus Kit [53] | SARS-CoV-2 in Wastewater | Lower RNA quality compared to direct isolation kit [53] | Faster than PEG precipitation. | Potential for membrane clogging or virus retention. |
| Virus Detection | RT-ddPCR [53] | SARS-CoV-2 RNA | Higher sensitivity and specificity than RT-qPCR [53] | Absolute quantification without standard curve; more resilient to inhibitors. | Higher cost per reaction than RT-qPCR. |
| Virus Detection | RT-qPCR [53] | SARS-CoV-2 RNA, Influenza A/B, RSV | Lower sensitivity than RT-ddPCR [53]; Sensitivities of 54-98% depending on virus and assay [41] | Widely available, high-throughput, cost-effective. | Requires standard curve for quantification; prone to inhibition. |
| Virus Detection | Viral Culture/Isolation [41] | RSV, Influenza Virus | Lower sensitivity (57% for RSV, 54% for Influenza) compared to molecular methods [41] | Allows for study of live, infectious virus. | Slow (days to weeks), requires specialized cell lines and facilities. |
| Virus Detection | Antigen Immunoassays [41] | RSV, Influenza Virus | Variable sensitivity (59% for Influenza, 82% for RSV) [41] | Rapid, simple to use, low cost. | Lower sensitivity, especially in adult populations [41]. |
Detailed Experimental Protocols
Protocol 1: Rapid Isolation of Cell-Associated Viral Particles via Deionized Water Lysis
This protocol is optimized for the release of cell-associated viral particles from persistently infected cell cultures, as demonstrated with the LCMV MX strain [52].
Principle: A hypotonic environment causes water to diffuse into the host cell, leading to swelling and eventual lysis, thereby releasing intracellular viral particles.
Materials:
- Persistently infected cell monolayer
- Sterile deionized water
- Phosphate-Buffered Saline (PBS), sterile
- Cell scraper
- Centrifuge and tubes
Procedure:
- Cell Harvesting: Remove the culture medium from the persistently infected cell monolayer. Wash the cell layer gently twice with sterile PBS to remove residual media and extracellular debris.
- Hypotonic Lysis: Add a sufficient volume of sterile deionized water to completely cover the cell monolayer (e.g., 1-2 mL for a T25 flask). Incubate at room temperature for 5-10 minutes. Observe the cells under a microscope for signs of swelling and lysis.
- Cell Dislodgement: Using a sterile cell scraper, gently but thoroughly scrape the lysed cells from the surface of the flask.
- Clarification: Transfer the cell lysate suspension to a sterile centrifuge tube. Centrifuge at 500 - 1,000 x g for 5-10 minutes to pellet large cellular debris.
- Collection: Carefully collect the supernatant, which now contains the released cell-associated viral particles. This supernatant can be used directly for downstream applications, such as infectivity assays or RNA extraction, or aliquoted and stored at -80°C.
Protocol 2: Virus Concentration and RNA Isolation from Complex Aqueous Samples
This protocol compares different approaches for processing wastewater for SARS-CoV-2 detection, with principles applicable to other complex samples containing viruses [53].
Principle: Viral particles are first concentrated and enriched from a large sample volume before RNA extraction, improving the detection limit for downstream molecular assays.
Materials:
- Aqueous sample (e.g., wastewater supernatant)
- Centrifuge and bottles
- 0.45 μm syringe filters (optional, see Note below)
- Polyethylene Glycol (PEG-8000) and NaCl (for PEG method)
- Vivaspin centrifugal concentrators (50 kDa MWCO) (for ultrafiltration method)
- Zymo Environ Water RNA Kit (for direct isolation method)
- NucleoSpin RNA Virus kit
Procedure: A. Sample Pre-processing 1. Debris Removal: Centrifuge the sample at 4,000 x g for 30 minutes at 4°C. 2. Filtration (Optional): Filter the supernatant through a 0.45 μm syringe filter. Note: One study found filtration to be counterproductive when using the Zymo Environ Water RNA Kit [53]. 3. Freezing Note: Avoid freezing samples before processing, as this significantly reduces RNA yield [53].
B. Virus Concentration & RNA Isolation (Choose ONE method) * Method B1: PEG Precipitation * Incubate supernatant with 8% PEG-8000 and 0.3 M NaCl overnight (~16 hours) at 4°C [53]. * Centrifuge at 10,000 x g for 120 minutes at 4°C to pellet the virus [53]. * Discard supernatant and resuspend the pellet in 500 μL of a suitable medium like Opti-MEM [53]. * Isolate RNA from the resuspended pellet using the NucleoSpin RNA Virus kit as per manufacturer's instructions [53]. * Method B2: Ultrafiltration * Load the supernatant onto a Vivaspin centrifugal concentrator. * Centrifuge at 4,000 x g at 4°C, repeating until the entire sample volume is concentrated to ~500 μL [53]. * Recover the concentrated sample. * Isolate RNA from the concentrate using the NucleoSpin RNA Virus kit [53]. * Method B3: Direct Isolation * Aliquot 1 mL of pre-processed sample into a tube. * Add 70 μL of the provided Water Concentrating Buffer from the Zymo Environ Water RNA Kit [53]. * Continue with the remainder of the manufacturer's protocol for RNA purification [53].
Workflow Visualization
Diagram 1: Virus Isolation and Detection Workflow
Diagram 2: Virus Detection Method Hierarchy
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Virus Isolation and Detection
| Item | Function/Application | Specific Examples / Notes |
|---|---|---|
| NucleoSpin RNA Virus Kit | Column-based isolation of viral RNA from cell-free samples like serum, plasma, or cell culture supernatant. | Used following virus concentration methods like PEG precipitation or ultrafiltration [53]. |
| Zymo Environ Water RNA Kit | Combined viral enrichment and RNA purification from large-volume, complex aqueous samples (e.g., wastewater). | Provides superior RNA quality compared to other methods; integrates concentration and extraction [53]. |
| Polyethylene Glycol (PEG-8000) | Precipitating and concentrating viral particles from large volume samples via incubation and centrifugation. | Used with NaCl; requires long incubation (overnight) [53]. |
| Vivaspin Centrifugal Concentrators | Ultrafiltration devices for rapid concentration of viral particles from sample supernatants based on molecular weight cut-off. | 50 kDa molecular weight cut-off is typical; faster than PEG precipitation [53]. |
| RT-ddPCR Supermix | Reagent mix for Reverse Transcription Droplet Digital PCR, enabling absolute quantification of viral load without a standard curve. | Offers higher sensitivity and specificity than RT-qPCR, ideal for low viral load samples [53]. |
| RT-qPCR Assay Kits | Kits for Reverse Transcription Quantitative PCR for detection and relative quantification of specific viral pathogens. | Target conserved viral genes (e.g., RdRp, E for SARS-CoV-2); widely used for high-throughput testing [41] [53]. |
| Cell Culture Media & Supplements | Maintenance of permissive cell lines essential for viral culture, propagation, and isolation. | Eagle's minimum essential medium is commonly used; specific cell lines (e.g., WI38, MRC-5) are required for different viruses [41] [54]. |
The accurate quantification of infectious viral particles is a cornerstone of virology research, playing a critical role in drug development, vaccine production, and pathogenesis studies. Within the broader context of cell culture methods for virus isolation, determining viral infectivity moves beyond mere detection to provide a functional measure of infectious units, which is essential for establishing infection parameters, evaluating antiviral agents, and standardizing research outputs [55]. The two principal techniques for quantifying infectious virus are the plaque assay and the 50% tissue culture infectious dose (TCID50) assay, both of which rely on the biological principle that a single infectious virus particle can initiate an infection in a susceptible cell culture system [56] [57]. These endpoint dilution methods enable researchers to translate observable cellular changes into precise, quantitative data, forming the foundation for reproducible and comparable results across the global research community.
Fundamental Principles
The plaque assay and the TCID50 assay, while both serving to quantify viral infectivity, are based on distinct readouts and mathematical approaches. The plaque assay is a direct counting method, operating on the principle that each plaque (a clear zone in a cell monolayer) results from a single infectious virus particle [56]. This direct one-to-one relationship makes it a highly intuitive measure of plaque-forming units (PFU). In contrast, the TCID50 assay is an indirect, probabilistic method that determines the dilution at which a virus sample infects 50% of inoculated cell cultures [58] [56]. It is based on the statistical concept that at the endpoint dilution, there is a 50% probability that a single infectious particle is present and will cause infection [56].
Comparative Analysis of Assay Characteristics
Table 1: Comparison between Plaque Assay and TCID50 Assay
| Feature | Plaque Assay | TCID50 Assay |
|---|---|---|
| Principle | Direct counting of infectious units [56] | Probabilistic endpoint determination [56] |
| Readout | Visible plaques (zones of cell lysis/death) [59] | Cytopathic effect (CPE) in individual wells [58] |
| Overlay Medium | Requires semi-solid overlay (e.g., agarose) to restrict virus spread [56] | Liquid medium; allows virus spread through the culture [56] |
| Format | Typically performed in plates, dishes, or wells [59] | Typically performed in 96-well plates with multiple replicates [58] |
| Calculation | PFU/mL = (Plaque count) / (Dilution factor × Inoculum volume) [56] |
Determined by Reed-Muench or Spearman-Kärber method [56] |
| Result | Plaque-forming units per mL (PFU/mL) [56] | 50% tissue culture infectious dose per mL (TCID50/mL) [58] |
| Sensitivity | Considered highly direct and accurate [57] | Can be more sensitive but also more variable [57] |
| Theoretical Relationship | 1 PFU ≈ 0.7 TCID50 [56] | 1 TCID50 ≈ 1.44 PFU (theoretical) [60] |
Detailed Experimental Protocols
TCID50 Assay Protocol
The following protocol, adapted from working BSL-3 procedures for SARS-CoV-2, provides a robust framework for determining virus titer in both tissue samples and viral stocks [58].
Materials and Reagents
- Cells: Permissive cell line (e.g., Vero-TMPRSS2 for SARS-CoV-2) [58]
- Plates: 96-well tissue culture plates for cell seeding; 96-well deep well plates for serial dilutions [58]
- Media: Cell culture growth medium and 2% media for dilutions [58]
- Other Reagents: Paraformaldehyde (PFA) for fixation, crystal violet for staining [58]
- Equipment: Multichannel pipette, biosafety cabinet, cell culture incubator [58]
Procedure
- Cell Seeding: The day before the assay, seed an appropriate cell line (e.g., Vero-TMPRSS2) in a 96-well plate at a density of 2x10⁴ cells/well in 100 µL of growth medium. On the day of the assay, check for uniform confluency [58].
- Sample Preparation:
- Thaw virus samples on ice.
- For tissue samples, centrifuge at 5,000 rpm for 5 minutes to aggregate tissue debris. The supernatant will be used for the titer determination [58].
- Serial Dilution:
- Prepare a 96-well deep well plate by adding 900 µL of 2% media to each well required for the dilution series.
- Add 100 µL of virus supernatant to the first row of the deep well plate.
- Using a multichannel pipette, perform serial dilutions by mixing the first row ~20 times, then transferring 100 µL to the next row. Repeat this process down the plate, creating a logarithmic dilution series [58].
- Inoculation and Incubation:
- Remove the media from the 96-well cell plate.
- Transfer 200 µL from each dilution in the deep well plate to the corresponding wells of the cell culture plate, using the number of replicates as planned (e.g., triplicate or quadruplicate) [58].
- Incubate the plate until cytopathic effect (CPE) is visible. The incubation time is virus-dependent (e.g., ~3 days for SARS-CoV-2 WA1, ~4 days for Omicron variants) [58].
- Fixation and Staining:
- After the incubation period, add 100 µL of 10% PFA to all wells to fix the cells and inactivate the virus. Incubate for at least 1 day [58].
- Remove the liquid and add 75 µL of 0.1% crystal violet stain per well. Incubate for 15 minutes.
- Remove the crystal violet and wash the plate thoroughly with tap water to reveal the CPE. Wells infected with virus will show less staining due to cell death [58].
- Data Analysis and Titer Calculation:
Plaque Assay Protocol
This protocol outlines the standard method for quantifying infectious virus via plaque formation, which remains the gold standard for viruses like Chikungunya virus [59].
Materials and Reagents
- Cells: Confluent monolayer of permissive cells (e.g., Vero cells).
- Overlay Medium: Semi-solid medium such as agarose or carboxymethyl cellulose to restrict virus diffusion.
- Staining Reagents: Crystal violet or neutral red.
- Plates: Multi-well plates (e.g., 24-well plates).
Procedure
- Cell Preparation: Seed cells to form a confluent monolayer in a multi-well plate by the day of infection.
- Virus Inoculation:
- Prepare serial 10-fold dilutions of the virus sample in a suitable buffer or maintenance medium.
- Remove the growth medium from the cell monolayer and inoculate each well with a specific volume (e.g., 100-200 µL) of each virus dilution. Ensure even coverage by tilting the plate.
- Incubate the plate for a defined adsorption period (e.g., 1 hour) at 37°C, with occasional rocking to prevent drying.
- Overlay Application:
- After the adsorption period, remove the inoculum.
- Carefully add a semi-solid overlay medium (e.g., warmed agarose mixed with 2x concentration maintenance medium) to each well. This confines virus spread to neighboring cells, leading to discrete plaque formation.
- Allow the overlay to solidify at room temperature, then return the plate to the incubator.
- Incubation and Plaque Development: Incubate the plates for the time required for plaques to become visible (typically several days, depending on the virus).
- Staining and Visualization:
- Once plaques are evident, fix the cells by adding a formalin solution (e.g., 10% formalin) on top of the overlay or after its removal.
- Remove the overlay and stain the cell monolayer with a visible dye like crystal violet. Intact cells will take up the stain, while plaques will appear as clear, unstained areas.
- Alternatively, use a vital stain like neutral red, which is taken up by live cells; plaques will appear as clear zones against a red background.
- Plaque Counting and Titer Calculation:
- Count the number of distinct, well-separated plaques at a dilution that yields a countable number (typically 10-100 plaques).
- Calculate the viral titer using the formula:
PFU/mL = (Number of plaques) / (Dilution factor × Volume of inoculum in mL)[56]. Example: If 32 plaques are counted at the 10⁻³ dilution and the inoculum volume was 0.5 mL, the titer is 32 / (10⁻³ × 0.5) = 6.4 × 10⁴ PFU/mL [56].
Data Analysis and Calculations
TCID50 Calculation Methods
Reed-Muench Method
This cumulative method provides a clear visualization of infection dynamics across the dilution series [56].
- Well Scoring: For each dilution, count the number of positive (CPE-present) and negative (CPE-absent) wells.
- Cumulative Sums:
- For each dilution, calculate the cumulative positive wells by adding the positives at that dilution and all higher concentrations (less dilute).
- Calculate the cumulative negative wells by adding the negatives at that dilution and all lower concentrations (more dilute).
- Infection Rate: Calculate the infection rate for each dilution:
Cumulative Positives / (Cumulative Positives + Cumulative Negatives). - Proportionate Distance (PD): Identify the two dilutions that bracket the 50% infection rate. Calculate the PD between them:
PD = (Infection Rate above 50% - 50%) / (Infection Rate above 50% - Infection Rate below 50%). - Log TCID50 and Titer:
Log(TCID50) = Log(Dilution above 50%) + (-PD × Log(Dilution Factor)).TCID50/mL = 10^(Log(TCID50)) × (1 / Inoculum Volume in mL)[56].
Spearman-Kärber Method
This method is mathematically simpler and relies on the total number of infected wells [56].
- Formula:
Log(TCID50) = Log(Lowest dilution with 100% CPE) + 0.5 - (Sum of % infected wells at all dilutions / 100). - The titer is then adjusted for the inoculum volume as in the Reed-Muench method [56].
Modern Computational Analysis
Traditional calculation methods are being supplemented and replaced by more accurate computational tools. The midSIN software uses Bayesian inference to analyze endpoint dilution assay data and outputs a result in Specific INfections (SIN) per mL [60]. This unit directly corresponds to the number of infections a sample will cause per mL, making it more intuitive for achieving a desired multiplicity of infection (MOI) than the TCID50 unit. Studies have shown that midSIN's estimates are more accurate and robust than the Reed-Muench and Spearman-Kärber approximations [60].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Materials for Viral Quantification Assays
| Item | Function/Application | Example/Specification |
|---|---|---|
| Permissive Cell Lines | Supports viral replication and CPE/plaque formation. | Vero E6, Vero-TMPRSS2 (SARS-CoV-2), MDCK (Influenza), A549 [58] [61] [55]. |
| Multi-well Plates | Platform for cell culture and virus inoculation. | 96-well plates (TCID50), 24-well/6-well plates (Plaque Assay) [58] [59]. |
| Semi-Solid Overlay | Restricts virus diffusion for plaque formation. | Agarose, Carboxymethyl cellulose [56]. |
| Fixatives and Stains | Visualize areas of infection (plaques or CPE). | Crystal Violet, Paraformaldehyde (PFA), Neutral Red [58] [59]. |
| Virus Transport Medium | Preserves virus viability during sample storage and transport. | Contains proteins and antibiotics [11]. |
Advanced Techniques and Automated Workflows
The field of viral quantification is evolving with the integration of automation and artificial intelligence to increase throughput, objectivity, and accuracy.
Automated Plaque Counting
Software such as plaQuest has been developed as a stand-alone tool for automated quantification of plaques from scanned images of multi-well plates [59]. It uses algorithms from the OpenCV library to detect and count plaques, significantly reducing the time and labor associated with manual counting while maintaining a strong correlation with human analyst counts [59].
AI-Powered CPE Detection
The DVICE (Detection of Virus-Induced Cytopathic Effect) framework represents a significant advancement. It uses a convolutional neural network (EfficientNet-B0) and transmitted light microscopy images to automatically detect virus-induced CPE in a label-free manner [61]. This AI-based approach has been validated for a wide range of viruses, including SARS-CoV-2, influenza, and adenovirus, and is suitable for applications like drug screening and serum neutralization assays. It can robustly measure CPE and even demonstrate virus class specificity [61].
Visualizing Workflows
The following diagrams illustrate the key procedural and analytical pathways for the major viral quantification assays.
TCID50 Assay Workflow
Plaque Assay Workflow
Endpoint Dilution Data Analysis Pathway
Solving Common Challenges: Contamination, Viability, and Protocol Optimization
Identifying and Preventing Viral Contamination in Cell Cultures (EBV, OvHV-2)
Cell culture techniques are indispensable in modern biomedical research, playing a critical role in disease modeling, drug screening, and vaccine production [6]. However, viral contamination poses a significant threat to the integrity of these systems, potentially compromising experimental results and bioprocess safety. Unlike microbial contamination, viral contamination presents unique challenges due to the difficulty in detection and the absence of effective treatments for infected cultures [6] [62]. Among the prevalent contaminating agents, Epstein-Barr virus (EBV) and Ovine Herpesvirus 2 (OvHV-2), both gammaherpesviruses, demand particular attention due to their high prevalence and potential to cause widespread issues in cell cultures across multiple species [6] [63]. This application note provides a detailed analysis of the impact of EBV and OvHV-2 contamination and outlines essential protocols for their identification and prevention within the context of virus isolation research.
Viral Contamination Profiles: EBV and OvHV-2
Understanding the distinct characteristics of these viruses is fundamental to developing effective contamination control strategies.
Table 1: Characteristics and Impact of EBV and OvHV-2 Contamination
| Feature | Epstein-Barr Virus (EBV) | Ovine Herpesvirus 2 (OvHV-2) |
|---|---|---|
| Prevalence | ~98% of the adult human population [6] [63] | Nearly all domestic sheep; infects over 33 animal species [6] [63] |
| Primary Host | Humans | Sheep |
| Concern in Cell Culture | Presence of latent and active forms can compromise biological products for therapy and prophylaxis [6] | Wide host range makes it a critical concern for cell cultures across various species [6] [63] |
| Commonly Affected Cell Lines | B-lymphoblastoid cell lines (B-LCLs), 293 human embryonic kidney cells [63] | Ovine peripheral blood lymphocytes [63] |
Prevention and Quality Control Strategies
A proactive approach is vital to prevent the introduction and spread of viral contaminants. The strategies can be categorized based on the research setting.
General Prevention for Research Laboratories
- Aseptic Technique: Implement strict handling procedures, controlled access to cell culture areas, and proper training [62].
- Routine Screening: Conduct regular testing for mycoplasma and other microbial contaminants using PCR, fluorescence staining, or ELISA-based methods [62].
- Cell Bank Validation: Perform regular authentication and testing of frozen cell stocks to prevent the propagation of latent contamination [62].
- Biosafety Equipment: Utilize biosafety cabinets and maintain surface disinfection protocols to control the lab environment [64].
Enhanced Control for GMP Manufacturing
- Closed Processing Systems: Employ single-use systems (SUS) to reduce risks from reusable equipment [62].
- Environmental Monitoring: Operate within HEPA-filtered cleanrooms with strict gowning procedures and continuous monitoring for particulates and microbial burden [62].
- Validated Sterilization: Implement and validate 0.1–0.2 µm filtration systems for media and buffer sterilization [62].
Detection and Identification Methods
Early and accurate identification of contamination is crucial. The preferred methods for EBV and OvHV-2 leverage molecular techniques due to their sensitivity and specificity.
Table 2: Preferred Detection Methods for EBV and OvHV-2
| Virus | Preferred Detection Methods | Key Application Notes |
|---|---|---|
| EBV | PCR [6] [63] | Detects EBV DNA with high sensitivity and specificity; can identify both active and latent forms [6]. |
| In situ Hybridization (ISH) [63] | Used to detect EBV-encoded small RNAs (EBERs) within cells. | |
| EBNA Detection (ELISA/Western Blot) [63] | Identifies Epstein-Barr Nuclear Antigen (EBNA) proteins to confirm infection. | |
| Southern Blot [63] | Differentiates between latent (episomal) and lytic (linear) forms of EBV DNA. | |
| OvHV-2 | PCR [63] | The primary tool for detection, given the challenges in culturing the virus in vitro. |
Viral Contamination Detection Workflow: This diagram outlines the decision pathway for identifying and characterizing viral contamination in cell cultures, from initial suspicion to final classification of the infection type.
Experimental Protocols
Protocol: EBV Preparation from B95-8 Cell Line
This protocol details the production of Epstein-Barr virus from the marmoset lymphoblastoid cell line B95-8, a known producer of infectious EBV particles [63] [65].
Research Reagent Solutions:
- RPMI 1640 Medium: Base nutrient medium for lymphocyte and lymphoblastoid cell culture [65].
- Fetal Bovine Serum (FBS): Provides essential growth factors, hormones, and lipids for cell proliferation; used at 15-20% concentration [65].
- Cyclosporin A: Immunosuppressant drug used at 200 ng/ml to inhibit T-cell activity, preventing them from killing newly infected B-cells during lymphocyte transformation [65].
- DMSO (Dimethyl Sulfoxide): Cryoprotectant agent used at 10% concentration for preserving cells in liquid nitrogen [65].
Procedure:
- Thawing: Rapidly thaw cryopreserved B95-8 cells by submersing the vial in pre-warmed sterile water (37°C). Plate the suspension into a 25 cm² flask with 10 ml of RPMI 1640 + 15% FBS [65].
- Initial Culture: After 24 hours, replace 50% of the medium to dilute the residual DMSO. Expand cells to a 75 cm² flask at day 2-3 as cell density increases [65].
- Scale-up: At day 5-7, further expand the culture to a 150 cm² flask with 200 ml of medium [65].
- Virus Harvest: At day 12-15, harvest the supernatant by centrifugation to remove cells. Critical: Do not add or remove medium between the expansion and harvest steps, as the EBV particles are released into the supernatant [65].
- Quality Control: Prescreen the supernatant for mycoplasma contamination using a detection system like Gen-Probe [65].
- Preparation and Storage: Dilute the supernatant 1:1 with RPMI 1640 + 20% FBS + 400 ng/ml cyclosporin. Filter-sterilize through a 0.22 µm filter, aliquot, and snap-freeze in liquid nitrogen for long-term storage [65].
Protocol: Lymphocyte Isolation and Transformation with EBV
This method is used to generate immortalized lymphoblastoid cell lines (LCLs) by infecting human B-lymphocytes with EBV [65].
Procedure:
- Blood Collection and Processing: Collect 10-20 ml of blood in ACD (yellow top) or sodium heparin (green top) vacutainer tubes. Keep samples at room temperature and process within 24-48 hours [65].
- Ficoll-Paque Separation: Dilute blood 1:2 with sterile PBS. Carefully underlay with Ficoll-Paque PLUS and centrifuge at 400 x g for 30-40 minutes [65].
- Lymphocyte Harvest: Collect the peripheral blood mononuclear cell (PBMC) interphase band. Wash cells twice with PBS (100 x g for 10 min) to remove platelets and residual Ficoll [65].
- EBV Infection: Resuspend 3-5 x 10⁶ lymphocytes in 3 ml of prepared EBV supernatant. Incubate overnight at 37°C, 5% CO₂ in an upright T-flask [65].
- Post-Infection Culture: After 24 hours, add 7 ml of transformation medium (RPMI 1640 + 20% FBS + 200 ng/ml cyclosporin) [65].
- Maintenance and Feeding: Begin feeding the cells after 5-7 days by letting cells settle, carefully removing 5 ml of old medium, and adding 5 ml of fresh growth medium. Note: Allowing the medium to become slightly acidic (pH 6.5-6.8) and avoiding disruption of cell clumps is beneficial for transformation [65].
- Establishment of Cell Line: Once transformation is achieved (typically within 2-3 weeks), the LCLs can be maintained in RPMI 1640 + 10-15% FBS without cyclosporin, keeping cell concentration between 4 x 10⁵ and 1 x 10⁶ cells/ml [65].
Lymphocyte Transformation Workflow: This diagram visualizes the key steps involved in creating an immortalized lymphoblastoid cell line (LCL) through Epstein-Barr virus (EBV) infection of primary human B-lymphocytes.
Consequences and Mechanisms of Contamination
Viral contamination can alter cellular physiology and compromise research data. For example, recent research has shown that the EBV-encoded tegument protein BRRF2 is secreted from infected cells via extracellular vesicles (EVs) [66]. These BRRF2-containing EVs can be internalized by recipient cells, such as macrophages, where BRRF2 protein binds to and inhibits the innate immune sensor cGAS (cyclic GMP-AMP synthase) [66]. This inhibition disrupts the cGAS-STING signaling pathway, a critical arm of the anti-viral innate immune response, thereby facilitating viral immune evasion [66]. Elevated levels of BRRF2-positive EVs in patient serum have been correlated with a diminished response to anti-PD-1 immunotherapy, highlighting the profound impact a viral contaminant can have on host cell biology and therapeutic outcomes [66].
Robust monitoring and prevention of EBV and OvHV-2 contamination are essential for ensuring the safety and reliability of cell culture systems in virus isolation research. The protocols and detection strategies outlined here provide a framework for maintaining cell culture integrity. As the field advances, the development of even more sensitive and rapid detection methodologies will be crucial to address existing gaps and further safeguard bioprocesses against these pervasive viral threats [6] [63].
Addressing Bacterial, Fungal, and Mycoplasma Contamination Issues
In the context of virus isolation research, maintaining pristine cell cultures is not merely a matter of good laboratory practice—it is a fundamental prerequisite for obtaining valid, reproducible data. Contamination represents one of the most persistent and devastating challenges in both research and large-scale bioprocessing, with the potential to compromise experimental outcomes, lead to erroneous conclusions, and waste invaluable resources [62]. For researchers dedicated to virus isolation, contamination takes on additional significance, as compromised cellular systems can alter viral susceptibility, replication dynamics, and pathogenicity findings.
The vulnerabilities are particularly acute in virus research, where even subtle contaminants can interfere with virus-host interactions. Bacterial and fungal contaminants often cause rapid culture demise, while mycoplasma contamination presents a more insidious threat, frequently escaping detection while altering cellular metabolism and gene expression [62] [67]. The consequences extend beyond mere inconvenience; contaminated viral stocks or infected cell lines can propagate errors across multiple experiments and collaborations, potentially invalidating months of dedicated work. Within drug development pipelines, where cell cultures serve as critical platforms for vaccine development and antiviral screening, contamination events can delay critical timelines and compromise patient safety [62] [68].
This application note provides comprehensive protocols and strategic guidance for addressing the most prevalent contamination challenges—bacterial, fungal, and mycoplasma—specifically tailored to the unique requirements of virus isolation research. By implementing robust detection methodologies and preventive strategies, researchers can safeguard their valuable cultures and ensure the integrity of their virological investigations.
Contamination Profiles: Characteristics, Detection, and Impact
Different classes of contaminants present distinct challenges for cell culture systems, particularly in virus isolation research. The table below summarizes the key characteristics, detection methods, and specific impacts on virological studies for bacterial, fungal, and mycoplasma contamination.
Table 1: Comparative Profiles of Major Cell Culture Contaminants
| Contaminant Type | Visual Indicators | Detection Methods | Impact on Virus Isolation Research |
|---|---|---|---|
| Bacterial | Cloudy/turbid medium; rapid pH change (yellow); possible odor [67] [69] | Light microscopy (motile particles); culture turbidity; pH indicators [67] | Rapid cell death prevents viral replication; alters cytokine profiles affecting viral susceptibility [62] |
| Fungal | Filamentous structures ("fuzzy"); visible colonies; changes in medium surface tension [67] | Microscopic identification of hyphae or budding cells; visual colony inspection [67] | Consumes nutrients needed for host cells and viruses; can overgrow cultures entirely [67] |
| Mycoplasma | No visual signs; subtle changes in cell growth rate/morphology; reduced transfection efficiency [62] [67] | PCR assays (gold standard); fluorescence staining; ELISA; specific mycoplasma detection kits [62] [67] [70] | Alters host cell gene expression and metabolism; modifies cytokine production; compromises viral host interaction studies [62] [67] |
Advanced Considerations for Virus Research
The challenges of mycoplasma contamination deserve particular emphasis in virology research. Studies indicate that a substantial proportion of cell cultures—up to >80% in some reports—may be infected with mycoplasma, often without the researcher's knowledge [70]. Unlike bacteria that cause rapid culture collapse, mycoplasma can persist covertly, inducing subtle but significant changes in cellular function that critically impact virological studies. These contaminants can compete for essential biosynthetic precursors, modify host cell surface receptors vital for viral entry, and trigger cytokine release that creates an artifactual antiviral state [67]. The resulting data may show altered viral replication kinetics or cell entry patterns that reflect the contaminated environment rather than authentic viral biology.
For virus isolation work specifically, the integrity of the host cell system is paramount. Research has demonstrated that some cell lines, such as Hep-2, exhibit particular susceptibility to certain pathogens like Mycoplasma pneumoniae, which can be exploited for isolation purposes but also underscores their vulnerability to contamination [71]. Furthermore, the trend toward using complex culture systems (3D cultures, co-cultures, and organoids) for virus research creates additional niches where contaminants can establish footholds away from conventional detection methods.
Detection Protocols and Methodologies
Implementing robust, routine detection protocols is essential for identifying contamination before it compromises virus isolation research. The following section provides detailed methodologies for detecting bacterial, fungal, and mycoplasma contaminants.
Mycoplasma Detection via PCR
Polymerase chain reaction (PCR) represents the gold standard for mycoplasma detection due to its high sensitivity, specificity, and rapid turnaround time [70]. Commercial kits such as the MycoScope PCR detection kit can detect fewer than 5 mycoplasma genomes per microliter of sample, with results available in less than 3 hours [70].
Table 2: Key Reagents for Mycoplasma PCR Detection
| Reagent/Material | Specification/Function |
|---|---|
| PCR Primers | Target 16S rRNA coding region for detection of all common cell culture mycoplasma species [70] |
| DNA Polymerase | Use high-fidelity enzymes compatible with the detection kit; multiple commercial options validated [70] |
| Sample Template | Cell culture supernatant (50-100 µL); DNA extraction recommended but direct testing possible [70] |
| Positive Control | Mycoplasma DNA for verifying assay performance and sensitivity |
| Agarose Gel | 1-2% for electrophoresis; distinct band at ~500bp indicates positive result [70] |
Protocol: Mycoplasma Detection by PCR
- Sample Collection: Collect 100 µL of cell culture supernatant from test cultures. Avoid excessive cell debris which can inhibit PCR.
- DNA Extraction (Recommended): Use commercial DNA extraction kits according to manufacturer instructions. Elute DNA in 50 µL elution buffer.
- PCR Master Mix Preparation: Prepare reaction mixture according to kit specifications. Typically includes: PCR buffer, dNTPs, specific primer set, DNA polymerase, and nuclease-free water.
- Reaction Setup: Combine 5 µL of template DNA with 20 µL of master mix. Include appropriate positive and negative controls.
- Thermocycling Conditions:
- Initial Denaturation: 95°C for 5 minutes
- 35-40 Cycles: Denaturation at 95°C for 30 seconds, Annealing at 55-60°C for 30 seconds, Extension at 72°C for 1 minute
- Final Extension: 72°C for 7 minutes
- Result Analysis: Separate PCR products by agarose gel electrophoresis (1.5-2% gel). Visualize under UV light; positive samples show distinct band at approximately 500bp [70].
Novel Detection Methods: UV Absorbance with Machine Learning
Emerging technologies offer promising alternatives for rapid contamination screening. Researchers have developed a method that combines UV absorbance spectroscopy with machine learning to provide label-free, noninvasive detection of microbial contamination in under 30 minutes [72].
This approach measures the unique ultraviolet light "fingerprints" of cell culture fluids, with machine learning algorithms trained to recognize patterns associated with contamination. The method is particularly valuable as a preliminary continuous safety test during critical manufacturing processes for cell therapy products, and shows promise for application in virus research where traditional sterility testing can take 7-14 days [72].
Protocol: Rapid Screening via UV Absorbance
- Sample Preparation: Collect 200 µL of cell culture medium in a UV-transparent microplate.
- Spectrum Acquisition: Measure UV absorbance across 200-300 nm range using a plate reader.
- Data Analysis: Process spectral data through validated machine learning algorithm trained on contaminated vs. sterile samples.
- Interpretation: Algorithm provides a definitive "yes/no" contamination assessment within 30 minutes [72].
Conventional Microbiological Methods
While molecular methods offer speed and sensitivity, traditional techniques remain valuable for certain applications.
Direct Microscopic Examination
- Sample Collection: Aseptically remove 100 µL of culture medium.
- Slide Preparation: Place drop on clean glass slide, apply coverslip.
- Microscopy: Examine under 400x magnification for motile bacteria (1-5 µm particles) or fungal structures (hyphae, spores) [67].
- Staining (if needed): Gram staining can differentiate bacterial types.
Culture Turbidity and pH Monitoring
- Visual Inspection: Daily examination for cloudiness or unexpected color changes.
- pH Assessment: Monitor phenol red indicator (yellow = acidic; purple = basic).
- Documentation: Record any deviations from expected growth patterns.
The following workflow diagram illustrates the integrated approach to contamination detection, combining routine monitoring with specific diagnostic methods:
Prevention and Control Strategies
Effective contamination management requires a proactive, multi-layered prevention strategy. The table below outlines essential reagents and materials that form the foundation of contamination control in cell culture laboratories focused on virus isolation.
Table 3: Essential Research Reagents for Contamination Control
| Reagent/Material | Function in Contamination Control | Application Notes for Virus Research |
|---|---|---|
| Certified Mycoplasma-Free Cell Lines | Starting material verified free of latent mycoplasma contamination [67] | Essential for creating master cell banks for virus propagation; validate upon receipt |
| Virus-Screened Sera | Fetal bovine serum (FBS) tested for adventitious viruses [67] [73] | Critical for preventing introduction of viral contaminants that could interfere with target virus |
| Antibiotic-Free Media | Maintains selective pressure without masking low-level contamination [67] | Use during routine culture; antibiotics may be justified during specific virus isolation steps |
| Validated Sterilization Filters | 0.1-0.2 µm pore size for sterilizing heat-labile solutions [62] | Use for media, reagents, and some viral stock preparations; validate for specific applications |
| HEPA-Filtered Biosafety Cabinets | Provides sterile workspace for culture manipulations [62] | Regular certification required; essential for working with pathogenic viruses |
| Environmental Monitoring Plates | Detects airborne microbial contamination in culture areas [68] | Place in incubators, near BSCs, and culture work areas; monitor monthly |
Comprehensive Prevention Framework
Aseptic Technique and Work Practices
- Single-Cell Line Handling: Process only one cell line at a time to prevent cross-contamination [67].
- Strict BSC Protocols: Maintain proper airflow, minimize turbulence, and disinfect all items entering the cabinet [62].
- Personal Hygiene: Proper gowning, gloves, and avoiding talking over open cultures reduces human-derived contamination [68].
Environmental and Equipment Controls
- Regular Incubator Decontamination: Clean CO₂ incubators weekly, including shelves, door gaskets, and water trays [67].
- HEPA Filtration: Ensure certified HEPA filters in biosafety cabinets and cleanroom environments [62].
- Validated Sterilization: Autoclave all reusable equipment and use sterile single-use consumables where possible [62].
Quality Assurance Measures
- Cell Line Authentication: Perform STR profiling every 6-12 months to verify cell line identity [67].
- Routine Mycoplasma Screening: Test cultures every 1-2 months using PCR-based methods [67] [70].
- Reagent Quality Control: Quarantine and test new cell lines and critical reagents before integration [67].
The strategic relationship between various prevention elements and their implementation timeline can be visualized as follows:
Contamination control in cell culture represents an ongoing challenge that requires diligence, systematic monitoring, and continuous education. For virus isolation research specifically, where cellular health directly influences virological outcomes, implementing the comprehensive detection and prevention strategies outlined in this document is essential for generating reliable, reproducible data. By adopting a layered defense approach—combining rigorous aseptic technique, environmental controls, validated reagents, and routine quality assessment—research laboratories can significantly reduce contamination events and safeguard their valuable virological studies. The protocols and methodologies presented here provide a framework for maintaining culture purity, thereby ensuring the integrity of virus isolation research and its critical applications in vaccine development, antiviral discovery, and fundamental virology.
Optimizing Cell Viability and Culture Conditions for Different Virus Types
Cell culture systems are foundational to virology research, enabling virus isolation, propagation, and the development of vaccines and therapeutics. The core challenge lies in the intricate and often virus-specific relationship between the host cell system and the viral pathogen. Optimizing cell viability and culture conditions is not a one-size-fits-all endeavor; it requires a meticulous understanding of how different cell lines, media components, and process parameters interact to support robust viral replication while maintaining cell health. This application note provides a structured framework and detailed protocols to guide researchers in tailoring cell culture environments for a diverse range of virus types, thereby enhancing the efficiency and reliability of virological studies and production workflows.
Critical Factors for Optimization
Successful virus cultivation hinges on the careful control of several interdependent factors. The selection of an appropriate cell line forms the bedrock of this process, as susceptibility to infection is dictated by the presence of specific viral receptors and the intracellular machinery necessary for replication. Beyond selection, precise management of culture parameters—including the timing of infection, the quantity of virus used, and the composition of the culture environment—is essential to maximize viral yield without compromising cell viability.
Cell Line Selection and Virus Susceptibility
The intrinsic properties of a cell line determine its permissiveness to viral infection. The following table summarizes optimized culture parameters for different cell line-virus pairs, as demonstrated in recent research.
Table 1: Cell Line Susceptibility and Optimized Culture Conditions for Virus Isolation and Production
| Virus | Cell Line | Key Application/Finding | Optimal Multiplicity of Infection (MOI) | Critical Culture Parameters | Reported Output/Performance |
|---|---|---|---|---|---|
| Foot-and-Mouth Disease Virus (FMDV), Serotypes O & A [74] | IB-IS-2 | Primary isolation of field viruses from clinical samples | Not Specified | Three consecutive passages; monitoring for Cytopathic Effect (CPE) | 76% isolation rate (38/50 samples); Mean titer: 10^3.4 TCID50/mL |
| BHK-21 | Large-scale virus culture for vaccine production | Not Specified | Three consecutive passages; monitoring for CPE | 24% isolation rate (12/50 samples); Mean titer: 10^4.2 TCID50/mL | |
| Cacipacoré Virus (CPCV) [75] | Vero CCL-81 | Plaque-forming unit (PFU) and focus-forming unit (FFU) assays | Via serial dilution | Overlay: 0.2-0.8% methylcellulose or agarose; Incubation: 3-6 days; Fixation: Methanol/Acetone | Successful protocol standardization for viral quantification |
| BHK CCL-10 | Plaque-forming unit (PFU) and focus-forming unit (FFU) assays | Via serial dilution | Overlay: 0.2-0.8% methylcellulose or agarose; Incubation: 3-6 days; Fixation: Methanol/Acetone | Successful protocol standardization for viral quantification | |
| Lentivirus (LV) for Immune Cell Transduction [76] | T Cells | Engineering for CAR/TCR expression | 1-5 (Titration Required) | Pre-activation (e.g., CD3/CD28); Cytokines (IL-2, IL-7, IL-15); Spinoculation | Typical transduction efficiency: 30-70%; Target VCN: <5 copies/cell |
| Natural Killer (NK) Cells | Engineering for enhanced cytotoxicity | 5-20 (Titration Required) | High-titer VSV-G pseudotyped vectors; Cytokines (e.g., IL-15) | Lower baseline efficiency; requires optimized vectors |
The data illustrates that a cell line ideal for initial virus isolation is not necessarily the best for high-titer production. For instance, while IB-IS-2 cells were superior for isolating FMDV field strains, BHK-21 cells, once infected, generated a higher viral titer, making them more suitable for vaccine production [74]. Similarly, the susceptibility of immune cells like T and NK cells to viral transduction (e.g., with Lentivirus) is highly dependent on their activation state and requires specific culture additives like cytokines [76].
Monitoring Cell Health and Viral Replication
Maintaining high cell viability is critical for producing high-quality viral stocks. The following workflow outlines a standardized process for establishing and monitoring viral cultures, from cell seeding to final harvest, integrating key quality control checkpoints.
Diagram 1: A generalized workflow for establishing and monitoring viral cultures, incorporating key quality control checkpoints for cell viability and process efficiency.
Key metrics for monitoring include:
- Viability Assessment: Employ trypan blue exclusion or, for more sensitive detection of apoptosis, flow cytometry with Annexin V/7-AAD staining [76].
- Transduction Efficiency: For engineered viral vectors, measure the percentage of cells expressing the transgene via flow cytometry. Control the Vector Copy Number (VCN) using droplet digital PCR (ddPCR), with clinical programs generally maintaining VCN below 5 copies per cell for safety [76].
- Cytopathic Effect (CPE): Observe cultures regularly under an inverted microscope for virus-specific morphological changes, such as cell rounding, syncytia formation, and lysis [74] [6].
Detailed Experimental Protocols
Protocol: Isolation and Titration of Field Virus Strains in Adherent Cell Lines
This protocol is adapted from studies on FMDV [74] and CPCV [75], providing a robust method for isolating and quantifying viruses from clinical samples.
Application: Primary isolation of viruses from clinical specimens (e.g., epithelial samples, tissue homogenates) and subsequent quantification of viral titer. Principle: Processed samples are inoculated onto permissive cell monolayers. Virus replication is detected by observing CPE or using immunostaining. The titer is determined by endpoint dilution (TCID50) or plaque assay.
Materials & Reagents:
- Cell Lines: BHK-21, IB-IS-2, Vero CCL-81, or other virus-susceptible lines [74] [75].
- Growth Medium: DMEM or EMEM supplemented with 10% Fetal Bovine Serum (FBS), L-glutamine, and penicillin/streptomycin.
- Maintenance Medium: As above, but with 2% FBS.
- Sample Transport Medium: PBS or specialized medium with antibiotics/antimycotics.
- Overlay Medium: For plaque assays, use 0.2-0.8% methylcellulose or low-melting-point agarose in maintenance medium [75].
- Fixative & Stain: Ice-cold methanol/acetone (1:1) for immunostaining, or 2% crystal violet in 10% ethanol for plaque assays [75].
- Antibodies: Primary pan-viral antibody (e.g., 4G2 for orthoflaviviruses) and HRP-conjugated secondary antibody for focus-forming assays [75].
Procedure:
- Cell Seeding: Seed appropriate cell lines in multi-well plates (e.g., 6-well or 96-well) and incubate until they form confluent, healthy monolayers (typically 24-48 hours).
- Sample Preparation: Homogenize and clarify clinical samples by low-speed centrifugation. Filter sterilize (0.45 µm) if necessary.
- Inoculation:
- Aspirate growth medium from cell monolayers.
- Wash monolayers gently with sterile PBS or serum-free medium.
- Inoculate wells with a defined volume of processed sample. Include negative control wells with maintenance medium only.
- Incubate for 1-2 hours at 37°C (or virus-specific temperature) to allow for viral adsorption. Gently rock the plates every 15-20 minutes.
- Post-Inoculation Incubation:
- Virus Detection and Titration:
- For TCID50: Use 96-well plates. Record CPE in each well after 3-5 days. Calculate the titer using the Spearman-Kärber or Reed-Muench method [74].
- For Plaque Assay: After incubation, remove overlay, fix cells with methanol/acetone or formalin, and stain with crystal violet. Count clear plaques.
- For Focus-Forming Assay: Fix with methanol/acetone, permeabilize, and incubate with primary and secondary antibodies. Develop with a precipitating substrate (e.g., TrueBlue) and count foci [75].
Troubleshooting:
- No CPE/Staining: Repassage the culture supernatant onto fresh cells for up to three blind passages [74]. Confirm cell line susceptibility.
- Low Titer: Optimize MOI, cell confluency at infection, and composition of the maintenance medium.
- Non-Specific Plaques/Foci: Include appropriate controls and optimize antibody concentrations.
Protocol: Viral Transduction of Immune Cells for Therapy
This protocol outlines key considerations for transducing human immune cells (e.g., T cells, NK cells) with viral vectors (e.g., Lentivirus, Gamma-retrovirus) for cell therapy manufacturing, based on current best practices [76].
Application: Genetic modification of immune cells to express therapeutic transgenes, such as Chimeric Antigen Receptors (CARs). Principle: Activated immune cells are exposed to viral vectors in the presence of enhancers to facilitate gene transfer, then expanded to therapeutic doses.
Materials & Reagents:
- Immune Cells: Primary T cells, NK cells, isolated from PBMCs.
- Activation Reagents: Anti-CD3/CD28 beads or antibodies.
- Cytokines: Recombinant human IL-2 (for T cells), IL-15 (for NK cells).
- Viral Vector: LV or γRV, high-titer, pseudotyped (e.g., VSV-G).
- Transduction Enhancers: Retronectin, protamine sulfate, or other commercial enhancers.
- Culture Medium: X-VIVO 15 or RPMI-1640, supplemented with 5-10% FBS or human serum.
Procedure:
- Cell Isolation and Activation: Isolate PBMCs and enrich for target cells. Activate T cells with anti-CD3/CD28 for 24-48 hours. This upregulates viral receptors and is critical for γRV transduction [76].
- Pre-Transduction Setup:
- Coat culture plates with retronectin (or other enhancer) if required.
- Centrifuge the required volume of viral vector onto the plate ("spinoculation" can enhance efficiency [76]).
- Transduction:
- Resuspend activated cells in fresh medium containing cytokines and the appropriate MOI (e.g., 1-5 for T cells, higher for NK cells [76]).
- Add the cell-vector suspension to the prepared plates.
- Centrifuge (e.g., 2000 × g, 90-120 minutes at 32°C) for spinoculation.
- Incubate for a further 6-24 hours.
- Post-Transduction Culture:
- Remove the vector-containing medium and replace with fresh growth medium with cytokines.
- Expand cells for several days, monitoring cell density, viability, and function.
- Harvest and Analysis:
- Harvest cells when the target cell number is achieved.
- Analyze transduction efficiency (flow cytometry for transgene expression), VCN (ddPCR), viability, and sterility [76].
Troubleshooting:
- Low Transduction Efficiency: Optimize MOI, cell activation status, and type/concentration of transduction enhancer. Use higher-titer vectors.
- Poor Cell Viability Post-Transduction: Titrate the MOI to reduce viral load toxicity. Ensure culture is supplemented with appropriate cytokines and avoid over-confluence.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Viral Culture and Transduction workflows
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Cell Lines | BHK-21, Vero CCL-81, A549, Calu-3, HEK-293T | Provide a permissive environment for viral replication; used for isolation, titration, and production [74] [75] [77]. |
| Culture Media & Supplements | DMEM, RPMI-1640, Fetal Bovine Serum (FBS), Penicillin-Streptomycin | Support cell growth and maintenance; serum concentration is often reduced during virus replication phase [75] [76]. |
| Viral Vectors & Transduction Aids | Lentivirus (VSV-G pseudotyped), Retronectin, Protamine Sulfate | Deliver genetic material to target cells (e.g., immune cells); enhancers increase vector/cell contact and transduction efficiency [76]. |
| Cell Activation & Cytokines | Anti-CD3/CD28 beads, Recombinant IL-2, IL-7, IL-15 | Prime immune cells for transduction and support their survival, expansion, and function post-transduction [76]. |
| Assay & Analysis Kits | NucleoSpin RNA/Plasmid kits, TRIzol, Flow Cytometry Antibodies, ddPCR Kits | Used for nucleic acid extraction, quantification of transduction efficiency, vector copy number, and cell phenotyping [76] [78]. |
| Overlay Materials | Methylcellulose, Low-Melting-Point Agarose | Restrict viral spread in plaque assays, enabling formation of discrete plaques for accurate quantification [75]. |
Within the broader research on cell culture methods for virus isolation, achieving high viral yields is a cornerstone for successful applications in vaccine development, virology research, and biotherapeutics. A common and significant challenge faced by researchers is the sudden or consistent occurrence of poor viral titers in otherwise standard protocols. This application note addresses this problem by dissecting two critical, often interrelated factors: culture medium selection and key culture parameters. The "cell density effect," a phenomenon where increasing cell density paradoxically reduces cell-specific viral yield, is a key focus [79]. Furthermore, the transition from poorly defined, serum-containing media to animal-component-free (ACF) or chemically defined media (CDM) is crucial for reducing variability, enhancing reproducibility, and mitigating contamination risks in manufacturing [79] [80]. The following sections provide a structured analysis of these challenges, supported by experimental data and detailed protocols, to equip researchers with strategies for troubleshooting and optimizing their viral production systems.
Core Challenges in Viral Production
The Cell Density Effect
The cell density effect describes the unexpected decrease in cell-specific viral yield observed as cell density increases in a bioreactor [79]. This is a major concern for process intensification, where high cell densities are often targeted to maximize volumetric productivity. Research on Foot-and-mouth disease virus (FMDV) production has quantitatively demonstrated this phenomenon.
Table 1: Impact of Cell Density and Media Exchange on FMDV Titer [79]
| Cell Density at Infection (cells/mL) | Media Condition | Viral Titer (TCID₅₀/mL) | Notes |
|---|---|---|---|
| 1 × 10⁶ | 30% Fresh Media | Baseline | |
| 2 × 10⁶ | 30% Fresh Media | Significant Reduction | Increased yield variability |
| 3 × 10⁶ | 30% Fresh Media | Lowest Titer | Highest yield variability |
| 1 × 10⁶ | 100% Media Exchange | Baseline | |
| 2 × 10⁶ | 100% Media Exchange | Slight Reduction | Mitigated yield variability |
| 3 × 10⁶ | 100% Media Exchange | Mitigated Reduction | Most consistent yields |
This effect is attributed to the cumulative impact of nutrient depletion and the accumulation of inhibitory metabolites (e.g., lactate and ammonia) in the spent medium when cells are grown to high densities [79]. The composition of the cell culture medium itself can influence the severity of this effect.
Serum-Based vs. Animal-Component-Free (ACF) and Chemically Defined Media (CDM)
The choice between traditional serum-based media and modern ACF/CDM involves a critical trade-off between cell growth support and process consistency.
- Serum-Based Media: Fetal Bovine Serum (FBS) provides a rich, albeit undefined, mixture of growth factors, hormones, and attachment factors that support robust cell growth and can stabilize viral particles [80]. However, it introduces significant challenges, including high lot-to-lot variability, risk of contamination with adventitious agents (e.g., viruses, prions), ethical concerns, and complexity in downstream purification [79] [80].
- ACF and CDM: These media offer a chemically consistent formulation that minimizes variability, enhances process control, and reduces regulatory concerns [79]. While early formulations sometimes struggled to match the performance of serum, modern ACF and CDM are increasingly optimized for specific cell lines and viral production processes. For instance, certain CDM prototypes (e.g., BHK300G) have been shown to mitigate the cell density effect better than others [79].
Table 2: Comparison of Media Types for Viral Production
| Media Type | Composition | Key Advantages | Key Disadvantages |
|---|---|---|---|
| Serum-Based | Complex, undefined mixture of growth factors, proteins, etc. | Supports growth of a wide range of cells; buffers and protects viral particles. | High lot-to-lot variability; risk of contamination; ethical issues; complicates downstream processing [80]. |
| Animal-Component-Free (ACF) | Free of animal-derived components; may still contain plant or synthetic hydrolysates. | Reduced variability and contamination risk compared to serum; more ethical. | Formulation may not be fully chemically defined; may require cell line adaptation [79]. |
| Chemically Defined (CDM) | Every component is a known chemical entity. | Maximum lot-to-lot consistency; simplified downstream processing; no animal-derived components. | Can be cell-line specific; may require supplements; development can be time-consuming and costly [79] [80]. |
Experimental Protocols for Optimization
Protocol: Mitigating Cell Density Effects via Media Exchange
This protocol is designed to evaluate and counteract the cell density effect in a spin tube system, adaptable to bioreactor scale [79].
1. Materials:
- Suspension cells (e.g., BHK-21)
- ACF or CDM (e.g., Cellvento BHK-200, BHK300G)
- Phosphate Buffered Saline (PBS), without calcium and magnesium
- Centrifuge
- TubeSpin bioreactors
- Shaker incubator
- FMDV (or virus of interest)
- Cell counter
2. Method:
- Inoculation and Growth: Inoculate TubeSpin bioreactors with initial viable cell densities of 0.5 × 10⁶, 1.0 × 10⁶, and 1.5 × 10⁶ cells/mL in a working volume of 30 mL.
- Incubation: Place cultures in a shaker incubator (e.g., 320 rpm, 37°C, 5% CO₂) overnight to reach target infection densities of 1 × 10⁶, 2 × 10⁶, and 3 × 10⁶ cells/mL, respectively.
- Media Exchange: Pellet cells at 290 × g for 5 minutes. For each target density, prepare two sets of tubes:
- Set A (100% Media Exchange): Completely remove and discard the spent medium. Resuspend the cell pellet in 30 mL of 100% fresh, pre-warmed medium.
- Set B (30% Media Supplement): Remove and discard 70% of the spent medium (21 mL). Resuspend the cell pellet in the remaining 9 mL of spent medium and add 9 mL of fresh medium (resulting in a final volume with 30% fresh medium).
- pH Adjustment: Adjust the pH of all cultures to 7.5 using 1M sodium hydroxide, if necessary.
- Infection: Infect all cultures with the virus at a low multiplicity of infection (MOI) of 0.01.
- Harvest: Incubate for 20 hours post-infection. Harvest the virus by centrifuging the culture at 3200 × g for 10 minutes at 4°C to remove cell debris. Store the supernatant (viral harvest) at -70°C.
- Titration: Determine the viral titer via an appropriate method, such as endpoint titration (TCID₅₀) on adherent indicator cells.
Workflow for Optimizing Media and Cell Density
The following diagram illustrates a logical workflow for troubleshooting poor viral yield by systematically investigating medium and cell density parameters.
Protocol: Adaptation to Serum-Free and Chemically Defined Media
Transitioning from serum-dependent to serum-free/CDM suspension culture is a key strategy for modernizing viral vector and vaccine production [80].
1. Materials:
- Adherent cell line (e.g., HEK293)
- Serum-containing medium (e.g., DMEM + 10% FBS)
- Target serum-free/CDM (e.g., FreeStyle 293, Ex-CELL 293)
- Non-enzymatic cell dissociation reagent (e.g., EDTA) or mild protease (e.g., TrypLE)
- Shaker flasks or bioreactors
2. Method:
- Initial Passage: Begin with adherent cells in the serum-containing medium. Passage the cells as usual using a dissociation reagent.
- Gradual Adaptation:
- Passage 1: Resuspend the cell pellet in a mixture of 75% serum-containing medium and 25% serum-free/CDM. Seed the cells into a standard culture vessel.
- Subsequent Passages: At each subsequent passage, gradually increase the proportion of serum-free/CDM (e.g., 50%/50%, 25%/75%) while decreasing the serum-containing medium.
- Final Stage: Once cells are growing in 100% serum-free/CDM, adaptation to suspension can begin. Seed the cells into a shaker flask with the serum-free/CDM and place them on an orbital shaker in a CO₂ incubator. Use a low agitation speed (e.g., 80-100 rpm) to start, gradually increasing it as cells adapt.
- Monitoring: Closely monitor cell viability, doubling time, and aggregate formation. Adaptation can take several weeks. Once stable growth in 100% serum-free suspension is achieved, the cell line is ready for optimized viral production studies.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Viral Production
| Item | Function | Example(s) |
|---|---|---|
| Chemically Defined Media (CDM) | Provides consistent, animal-component-free nutrients for cell growth and viral replication. Reduces lot-to-lot variability. | BHK300G [79], BalanCD HEK293 [80] |
| Animal-Component-Free (ACF) Media | Supports cell growth without animal-derived components, reducing contamination risk while potentially offering richer nutrition than basic CDM. | Cellvento BHK-200 [79] |
| Serum-Free Media (SFM) | Formulated for specific cell types without serum; may still contain plant-derived hydrolysates. | FreeStyle F17, VP-SFM [80] |
| Cell Dissociation Reagents | Detaches adherent cells for passaging or infection. Non-enzymatic or mild enzymatic reagents help preserve surface proteins. | TrypLE, Accutase, EDTA [64] [81] |
| Antifoaming Agents | Controls foam formation in bioreactors, which is crucial for maintaining gas transfer and cell viability in high-density cultures. | Animal Origin-Free (AOF) Antifoam [79] |
| Supplements (e.g., ITS) | Insulin-Transferrin-Selenium supplement provides key growth-promoting factors in a chemically defined format. | ITS Supplement [80] |
| Cell Retention Devices | Enables continuous perfusion cultures by retaining cells in the bioreactor while removing spent media, essential for mitigating cell density effects. | Alternating Tangential Flow (ATF) systems, Acoustic Filters [82] |
Successfully troubleshooting poor viral yield requires a systematic approach that focuses on the critical interplay between medium composition and culture parameters. The cell density effect is a major, yet addressable, obstacle to process intensification. A complete media exchange before infection is a highly effective strategy to counteract this phenomenon by replenishing nutrients and removing inhibitors [79]. Furthermore, the transition from serum-based to ACF or CDM, while potentially challenging, is a necessary evolution for achieving a robust, scalable, and compliant manufacturing process for viruses and viral vectors [79] [80]. By applying the diagnostic workflows, experimental protocols, and reagent knowledge outlined in this application note, researchers can make informed decisions to optimize their systems and ensure the consistent production of high-titer viral stocks.
The isolation and purification of biological nanoparticles, including viruses and extracellular vesicles (EVs), are critical steps in biomedical research, diagnostics, and therapeutic development. Among the various techniques available, ultracentrifugation, filtration, and precipitation have emerged as foundational methods for obtaining high-purity preparations from complex biological matrices. These techniques enable researchers to separate target particles based on their physical properties including size, density, and solubility [83] [84].
Within the context of virus isolation research, particularly using cell culture systems, selecting the appropriate purification strategy directly impacts multiple downstream applications. The purity, integrity, and biological activity of isolated viral particles or EVs can influence experimental outcomes in drug development, vaccine production, and gene therapy vector characterization [85] [86] [87]. This article provides detailed application notes and protocols for implementing these key purification techniques within a virology research framework.
Core Principles of Key Purification Techniques
Ultracentrifugation
Ultracentrifugation employs high centrifugal forces to separate particles based on their density and sedimentation coefficients. As the current gold standard for virus and EV purification, this technique leverages high-speed centrifugation (typically 100,000-110,000 ×g) to pellet nanoparticles against solution viscosity and Brownian motion [83] [84]. The process typically involves multiple steps of differential centrifugation to first remove larger debris and dead cells at lower speeds (300-10,000 ×g) followed by high-speed pelleting of the target particles [83]. Ultracentrifugation is particularly valued for preserving biological activity and providing high-purity isolates suitable for proteomic and glycomic analysis [84].
Filtration-Based Methods
Filtration techniques separate particles based primarily on size exclusion using membranes with defined pore sizes. Ultrafiltration employs porous membranes or centrifugal filters with specific molecular weight cut-offs (MWCO) to concentrate viral particles or EVs while allowing smaller soluble components to pass through [83]. Common configurations include Amicon filters with MWCO of 10 kDa or 100 kDa, which effectively retain particles larger than approximately 30 nm [83]. The choice of membrane material (regenerated cellulose, polyethersulfone, cellulose triacetate, or Hydrosart) impacts recovery efficiency and potential for non-specific binding [83].
Precipitation Methods
Precipitation techniques reduce the solubility of target particles in solution, facilitating their isolation by low-speed centrifugation. Polymer-based precipitation, commonly using polyethylene glycol (PEG), works by excluding volume around particles, effectively reducing their solubility [83]. Charge-based precipitation utilizes positively charged molecules like protamine to interact with negatively charged particles surfaces [83]. Organic solvent precipitation approaches, such as the Protein Organic Solvent Precipitation (PROSPR) method, remove soluble proteins while leaving lipid-encapsulated particles in solution [83].
Comparative Performance Analysis
Quantitative Comparison of Isolation Techniques
Table 1: Comparative analysis of ultracentrifugation, filtration, and precipitation methods for nanoparticle isolation
| Parameter | Ultracentrifugation | Ultrafiltration | Precipitation |
|---|---|---|---|
| Mean Particle Size | 60 nm [83] | 122 nm [83] | 89 nm [83] |
| Size Distribution | Narrow [83] | High variability [83] | Moderate [83] |
| Cell Viability Improvement | 22% [83] | 11% [83] | 15% [83] |
| Live Cell Content (Flow Cytometry) | 20% increase [83] | 9% increase [83] | 15% increase [83] |
| Equipment Requirements | Ultracentrifuge [83] | Centrifugal filters [83] | Standard centrifuge [83] |
| Processing Time | 70-120 min (plus additional cleaning steps) [83] | Time-consuming [83] | Overnight incubation [83] |
| Scalability | Moderate [87] | High | High |
| Cost Considerations | High equipment cost [87] | Moderate | Low |
Table 2: Applications in virus and extracellular vesicle research
| Method | Best Applications | Limitations | Suitability for Downstream Analysis |
|---|---|---|---|
| Ultracentrifugation | Proteomic & glycomic studies [84]; Gene therapy vector purification [87]; Fundamental virology research | High equipment cost [87]; Potential for particle damage [84] | Excellent for proteomics, glycomics, functional studies [84] |
| Ultrafiltration | Rapid concentration; Serum-free applications; Large volume processing | Clogging issues; Membrane adsorption losses; Size heterogeneity in isolates [83] | Good for molecular analysis; Potential protein contamination |
| Precipitation | High-throughput screening; Diagnostic biomarker discovery; Large sample processing | Co-precipitation of contaminants; Requires additional cleaning steps; Polymer contamination [83] | Moderate; may require additional purification steps |
Technique Selection Guidelines
The optimal purification method depends on specific research objectives and technical constraints. Ultracentrifugation remains the preferred choice for applications requiring high purity and preservation of biological activity, particularly for sensitive downstream applications like proteomic analysis [84]. Filtration methods offer advantages when processing large volumes or when specialized equipment is unavailable. Precipitation techniques provide accessibility and scalability for high-throughput applications where absolute purity may be less critical [83].
For virus purification specifically, ultracentrifugation enables separation of empty versus full capsids—a critical quality attribute for gene therapy vectors like AAV [87]. Continuous ultracentrifugation presents a scalable solution for industrial-scale virus capture, concentration, and separation of different capsid populations, though infrastructure costs remain a limitation [87].
Detailed Experimental Protocols
Protocol 1: Ultracentrifugation for Virus/EV Isolation from Cell Culture
This protocol outlines the standard ultracentrifugation procedure for isolating viral particles or extracellular vesicles from cell culture supernatants, adapted from methodologies used in EV and virus research [83] [84] [86].
Materials:
- Pre-cleared cell culture supernatant
- Ultracentrifuge with fixed-angle or swinging-bucket rotor
- Polycarbonate bottles or open-top thin-walled ultracentrifuge tubes
- Phosphate-buffered saline (PBS), ice-cold
- Sterile resuspension buffer
Procedure:
- Sample Preparation: Begin with conditioned cell culture media from virus-infected or EV-producing cells. For EV isolation, use media supplemented with exosome-depleted FBS [83].
- Clarification Centrifugation: Transfer supernatant to centrifuge tubes and spin at 300 ×g for 10 minutes at 4°C to remove dead cells [83]. Collect supernatant without disturbing the pellet.
- Debris Removal: Centrifuge the resulting supernatant at 10,000 ×g for 30 minutes at 4°C to eliminate larger vesicles and cellular debris [84].
- Ultracentrifugation: Transfer the clarified supernatant to ultracentrifuge tubes. Balance tubes precisely and centrifuge at 100,000 ×g for 70-120 minutes at 4°C [83].
- Pellet Washing: Carefully decant the supernatant and resuspend the pellet in a suitable volume of ice-cold PBS (typically 1-5 mL depending on initial volume) [83].
- Second Ultracentrifugation: Repeat the ultracentrifugation step at 100,000 ×g for 70 minutes at 4°C to improve purity [83].
- Final Resuspension: Discard the supernatant and resuspend the final pellet in an appropriate storage buffer (e.g., PBS or specialized cryopreservation buffer). Aliquot and store at -80°C [84].
Technical Notes:
- Maintain samples at 4°C throughout the procedure to minimize degradation
- Use protease inhibitors if subsequent protein analysis is planned
- For larger volumes, consider continuous ultracentrifugation systems [87]
- Optimal g-force and duration may require optimization for specific particle types
Protocol 2: Ultrafiltration for Virus/EV Concentration
This protocol describes concentration of viral particles or EVs using size-based ultrafiltration membranes [83].
Materials:
- Clarified cell culture supernatant
- Ultrafiltration devices (e.g., Amicon Ultra with 10-100 kDa MWCO)
- Standard laboratory centrifuge
- Phosphate-buffered saline (PBS)
Procedure:
- Filter Selection: Choose an appropriate ultrafiltration device based on target particle size. For most viruses and EVs, 100 kDa MWCO membranes are suitable [83].
- Sample Loading: Transfer clarified supernatant to the filtration device according to manufacturer's volume specifications.
- Concentration: Centrifuge at the recommended g-force (typically 3,000-4,000 ×g) until the desired concentration factor is achieved.
- Buffer Exchange (Optional): Add PBS to the concentrated retentate and repeat centrifugation to replace the original medium with a more suitable buffer.
- Recovery: Invert the filtration device in a clean collection tube and centrifuge at 1,000 ×g for 2-3 minutes to recover the concentrated sample.
Technical Notes:
- Regenerated cellulose membranes with 10 kDa pore size show highest recovery efficiency for EVs [83]
- Avoid over-concentrating as this may increase particle aggregation
- Different membrane materials (regenerated cellulose, polyethersulfone, cellulose triacetate) yield different recovery efficiencies [83]
Protocol 3: Polymer-Based Precipitation for Virus/EV Isolation
This protocol outlines precipitation-based isolation using polymers such as polyethylene glycol (PEG) [83].
Materials:
- Cell culture supernatant
- Precipitation solution (commercial kits or laboratory-prepared PEG solution)
- Standard laboratory centrifuge
- Phosphate-buffered saline (PBS)
Procedure:
- Sample Preparation: Clarify cell culture supernatant by centrifugation at 2,000 ×g for 30 minutes to remove cells and debris.
- Precipitation: Mix the clarified supernatant with precipitation solution according to manufacturer's instructions (typically 1:2 to 1:5 ratio).
- Incubation: Incubate the mixture overnight at 4°C to allow complete precipitation.
- Recovery: Centrifuge the sample at 10,000 ×g for 60 minutes at 4°C to pellet the precipitated particles.
- Resuspension: Discard the supernatant and resuspend the pellet in PBS or an appropriate buffer for downstream applications.
Technical Notes:
- Precipitation efficiency depends on polymer concentration, molecular weight, and incubation time
- Isolates may contain co-precipitated proteins and require additional cleaning steps for high-purity applications
- Commercial precipitation kits are available that optimize polymer composition and ratios
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential research reagents and materials for purification workflows
| Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Cell Culture Media | DMEM, RPMI-1640 with 10% FBS [85] [84] | Cell maintenance and virus/EV production; Use exosome-depleted FBS for EV studies [83] |
| Centrifugation Equipment | Ultracentrifuge with fixed-angle rotors [83]; Standard benchtop centrifuges | Particle pelleting; Differential centrifugation requires multiple rotor systems |
| Filtration Devices | Amicon Ultra-2 (10-100 kDa MWCO) [83]; Vivaspin series; 0.22 μm sterilization filters [85] | Size-based separation and concentration; Sample sterilization and clarification |
| Precipitation Reagents | Polyethylene glycol (PEG) solutions [83]; Commercial kits (e.g., Total Exosome Isolation reagent) | Volume exclusion-based precipitation; Simplified workflow for rapid isolation |
| Chromatography Materials | Affinity resins; Ion exchange matrices; Size exclusion columns [87] | High-resolution purification; Empty-full capsid separation for AAV vectors [87] |
| Buffers & Solutions | Phosphate-buffered saline (PBS) [83]; Protease inhibitor cocktails; Density gradient media | Sample preservation; Maintaining physiological conditions; Enhanced separation |
| Characterization Tools | Transmission electron microscopy [84]; Dynamic light scattering [83]; Nanoparticle tracking analysis | Size and morphology assessment; Concentration quantification; Purity evaluation |
Ultracentrifugation, filtration, and precipitation methods each offer distinct advantages and limitations for purifying viruses and extracellular vesicles from cell culture systems. The selection of an appropriate technique must consider research objectives, required purity, downstream applications, and available resources. Ultracentrifugation remains the gold standard for high-purity isolates, while filtration and precipitation offer practical alternatives for specific applications. As the field advances, particularly in gene therapy where AAV purification presents unique challenges [87], continued refinement of these core techniques will enhance their efficiency, scalability, and accessibility for research and therapeutic development.
Ensuring Accuracy: Validation Methods and Comparative Analysis of Techniques
Within the landscape of viral diagnostics and research, cell culture maintains its status as the gold standard for virus isolation and identification [5]. Despite the rapid ascent of molecular methods, which offer unparalleled speed and sensitivity for detecting viral genetic material, they cannot on their own confirm the presence of a viable, replicating pathogen [88] [89]. This Application Note delineates the indispensable role of cell culture methods in virology, framed within a broader thesis on their enduring value. It provides detailed protocols and quantitative data to guide researchers and drug development professionals in the application of these foundational techniques for isolating infectious viruses, a critical step in vaccine development, antiviral testing, and pathogen discovery [5] [89].
The Unmatched Value of Viral Viability Assessment
The principal strength of cell culture lies in its ability to confirm viral infectivity and replication, providing a complete picture of pathogen viability that molecular methods can only infer.
Direct Evidence of Infectivity: Molecular techniques, such as RT-PCR, detect the presence of viral genomic RNA (gRNA) but cannot distinguish between infectious virions and non-infectious viral debris [88]. This is a significant limitation for clinical management decisions, such as determining patient isolation periods or assessing the efficacy of disinfection processes [90]. Cell culture directly demonstrates the presence of a functional, replicating virus by observing its ability to infect and propagate within a living host cell system [88].
Surrogate Markers and Their Limitations: To address the limitations of gRNA detection, markers like subgenomic RNA (sgRNA) have been investigated as proxies for active viral replication. A 2025 study demonstrated that while sgRNA detection showed high accuracy (98%) in predicting cell culture positivity, cell culture itself remains the definitive reference against which these surrogates are measured [88]. The study concluded that sgRNA is a superior marker to gRNA Ct values, but it still functions as a surrogate for the gold standard [88].
Essential for Public Health and Industry: In the water industry, regulatory requirements for virus removal are based on logs of infectious human pathogenic viruses. PCR-based methods are insufficient for this purpose, as they may overestimate the risk from non-infectious viral particles, particularly after disinfection [90]. Cell culture is therefore critical for validating the performance of water treatment processes.
The table below summarizes a comparative analysis of viability markers for SARS-CoV-2, illustrating the performance of various methods against the cell culture gold standard.
Table 1: Comparison of SARS-CoV-2 Viability Markers Using Cell Culture as Gold Standard
| Detection Method | Sensitivity | Specificity | Positive Predictive Value (PPV) | Negative Predictive Value (NPV) | Accuracy |
|---|---|---|---|---|---|
| gRNA RT-PCR (Ct ≤ 25) | 0.88 | 0.89 | 0.92 | 0.84 | 0.88 |
| gRNA RT-PCR (Ct ≤ 30) | ~1.0 | 0.24 | 0.63 | ~1.0 | Not Reported |
| sgRNA RT-PCR (E gene) | 0.99 | 0.96 | 0.97 | 0.99 | 0.98 |
Data adapted from a prospective study on immunocompromised patients (n=285 samples) [88].
Evolution of Cell Culture Methodologies
Cell culture techniques have evolved significantly from traditional methods, incorporating innovations that enhance speed, sensitivity, and convenience.
Traditional Cell Culture
The traditional method involves inoculating a clinical sample onto a monolayer of cells grown in a standard screw-cap tube. The culture is then incubated and monitored daily for a cytopathic effect (CPE), which refers to virus-induced morphological changes in the host cells [5]. These changes can include cell rounding, shrinking, swelling, or syncytium formation. A significant drawback of this method is the time to result, which can range from 1 day to several weeks depending on the virus [5]. For instance, Herpes Simplex Virus (HSV) may produce CPE in 24 hours, whereas Cytomegalovirus (CMV) can require 10-30 days [5].
Advanced Modern Formats
Modern approaches have streamlined the process to overcome the limitations of traditional culture.
Shell Vial / Microwell Culture: The traditional tube has been largely supplanted by smaller formats like the shell vial or microwell plates (e.g., 24-well clusters) [5] [89]. These formats are compatible with centrifuge-enhanced inoculation, where low-speed centrifugation forces the virus into contact with the monolayer, drastically reducing the absorption time and accelerating the entire detection process [89].
Cryopreserved Cell Culture: Commercially prepared, cryopreserved cell monolayers in ready-to-use vials offer significant convenience. These vials are stored in liquid nitrogen and can be rapidly thawed in a water bath for immediate sample inoculation, simplifying workflow and standardizing materials [5].
Cocultured and Transgenic Cell Lines: To broaden detection capabilities, mixed cell lines (e.g., R-Mix, a combination of A549 and mink lung cells) are used in a single vial to support the growth of a wide spectrum of viral pathogens [5]. Furthermore, transgenic cell lines are engineered with reporter genes that activate only in the presence of a specific virus, allowing for detection before CPE is visible and providing greater specificity [89].
The following diagram illustrates the core workflow and key decision points in a modern cell culture assay for virus isolation.
Detailed Experimental Protocol for Virus Isolation in Cell Culture
This protocol provides a step-by-step methodology for isolating viruses using a shell vial format with pre-CPE detection, balancing speed and reliability [5] [13].
Sample Collection and Processing
- Collection: Collect appropriate clinical specimens (e.g., nasopharyngeal swabs) in viral transport medium [88]. Transport to the laboratory on ice or at 4°C.
- Processing: Vortex the sample medium and discard the swab. Centrifuge the liquid medium to pellet debris, cells, and bacteria. Retain the supernatant, which contains the virus [5].
- Inoculum Preparation: Aseptically transfer 0.2-0.3 mL of the clarified supernatant to a shell vial containing a cryopreserved or freshly prepared cell monolayer (e.g., MRC-5, A549, or Vero E6 cells, chosen based on the suspected virus) [5] [13].
Virus Inoculation and Absorption
- Centrifuge Enhancement: Securely cap the shell vial and centrifuge at 700 x g for 45-60 minutes at room temperature. This step forces viral particles into contact with the cell monolayer, enhancing infection efficiency [89].
- Post-Inoculation Incubation: After centrifugation, carefully remove the inoculum supernatant. Wash the monolayer once with phosphate-buffered saline (PBS) to remove any residual inhibitors. Replace with fresh virus growth medium (typically supplemented with 2% fetal bovine serum) [13].
- Incubation: Incubate the culture at 35°C in a 5% CO₂ atmosphere for 24-48 hours, or as required for the target virus [5].
Virus Detection and Identification
- Pre-CPE Detection (Immunofluorescence): After the incubation period, this rapid method can be employed.
- Aspirate the medium from the vial.
- Fix the cell monolayer on the coverslip with cold acetone or methanol for 10 minutes.
- Add a virus-specific, fluorescein-labeled monoclonal antibody to the fixed cells.
- Incubate, then wash to remove unbound antibody.
- Examine the coverslip under a fluorescence microscope. Apple-green fluorescence indicates a positive infection, confirming virus isolation often before CPE is visible [5] [89].
- CPE-Based Detection: For traditional culture, examine the cell monolayer daily using an inverted microscope for signs of CPE. The time to CPE is virus-dependent [5].
- Confirmation: Positive cultures, whether identified by CPE or pre-CPE methods, should be confirmed by subpassaging or further molecular analysis to identify the virus type.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Virus Cell Culture
| Reagent / Material | Function / Application | Examples & Notes |
|---|---|---|
| Cell Lines | Propagation host for viral replication. | Vero E6 (SARS-CoV-2), MRC-5 (CMV, HSV), A549 (Adenovirus), RhMK (Influenza) [5] [88] [13]. |
| Growth Media | Supports cell viability and virus propagation. | Eagle's Minimum Essential Medium (EMEM), RPMI-1640; typically with 2-10% FBS for virus growth [13]. |
| Shell Vials / Cluster Plates | Container for growing cell monolayers. | Enables centrifuge-enhanced inoculation and easy microscopic examination [5] [89]. |
| Virus-Specific Antibodies | Detection and identification of isolated viruses. | Used in immunofluorescence assays for pre-CPE diagnosis; e.g., cocktail antibodies for respiratory viruses [5] [89]. |
| Cryopreservation Agents | Long-term storage of cell stocks and seed viruses. | Dimethyl sulfoxide (DMSO); storage in liquid nitrogen vapor phase (< -120°C) [5] [13]. |
Cell culture remains the cornerstone of virology for a simple, definitive reason: it confirms the presence of a replicating, infectious agent [5] [88]. While molecular methods provide critical speed for initial diagnostics, they answer a different question—"Is viral genetic material present?"—rather than "Is there an infectious virus?" [90] [89]. The continued innovation in cell culture formats, from shell vials to transgenic cells, ensures its relevance by addressing historical limitations of time and labor. For applications ranging from clinical management of immunocompromised patients and public health surveillance to fundamental virology research and drug discovery, the demonstration of viral viability via cell culture is an irreplaceable component of the scientific toolkit.
Cell culture is a cornerstone technique in virology, essential for virus isolation, vaccine production, and fundamental research into viral pathogenesis. The methodology has evolved significantly from its origins, transitioning from traditional formats to sophisticated modern systems that offer enhanced sensitivity, speed, and specificity. This evolution reflects the continuous pursuit of more physiologically relevant and experimentally efficient models to study viral behavior and host-pathogen interactions. Within the context of a broader thesis on cell culture methods for virus isolation research, this analysis provides a critical examination of both traditional and contemporary cell culture platforms, detailing their technical specifications, applications, and limitations. The shift from traditional to modern formats represents a paradigm change in how researchers approach virus isolation, balancing the need for robust, gold-standard methods with the demand for rapid, high-throughput diagnostics suitable for both research and clinical applications [5] [1]. This document provides a detailed comparative analysis structured for researchers, scientists, and drug development professionals, incorporating specific protocols and reagent solutions to facilitate practical implementation in the laboratory.
Traditional Cell Culture Formats: Foundations and Methodologies
Traditional cell culture methods have served as the foundation for virology for decades. The standard container for traditional culture has been the screw-cap tube glass (16 mm × 125 mm), in which monolayer cells grow on one side of the glass surface [5] [1]. For accurate virus identification, laboratories must maintain multiple cell line types, with the most common being primary rhesus monkey kidney cells (RhMK), primary rabbit kidney cells, MRC-5 (human lung fibroblast), human foreskin fibroblasts, HEp-2 (human laryngeal carcinoma), and A549 (human lung carcinoma) [5]. The cost for traditional cell culture ranges from approximately $1.5 to $6.50 per tube, with the final expense dependent on the number and types of cell lines required for comprehensive viral diagnosis [5] [1].
The fundamental principle of virus detection in traditional culture relies on observing virus-induced morphological changes in the host cell monolayer, known as the cytopathic effect (CPE). CPE manifests as visible alterations in cell morphology—including cell rounding, shrinking, swelling, or syncytium formation—that indicate viral replication and propagation [5] [6]. The time required for CPE appearance varies significantly among viruses, from as little as 24 hours for Herpes Simplex Virus (HSV) to 10-30 days for Cytomegalovirus (CMV), with most viruses requiring 5-10 days of incubation [5]. While CPE observation provides preliminary virus identification, confirmatory testing such as immunofluorescence (IF) assay is typically required for definitive viral typing [5].
Detailed Protocol: Traditional Tube Cell Culture for Virus Isolation
- Sample Preparation: Vortex the sample medium and discard the swab. Centrifuge the liquid medium to pellet fungi, cells, bacteria, and blood components. Retain the supernatant, which contains viral particles [5].
- Cell Inoculation: Aspirate 0.2–0.3 mL of the processed supernatant and add it to the cell culture tube. Incubate at 35°C with 5% CO₂ for 90 minutes to facilitate viral adsorption to the monolayer [5].
- Maintenance and Observation: Discard the inoculum and replace it with fresh maintenance medium. Return the culture to the incubator. Examine the cell monolayer daily using an inverted microscope for development of CPE [5] [89].
- Virus Identification and Confirmation: Document the type of CPE observed and correlate with the cell line used and incubation period. For definitive identification, perform confirmatory tests such as immunofluorescence with virus-specific antibodies [5].
Characteristic CPE Patterns of Common Viruses
The table below summarizes the characteristic cytopathic effects of common viruses in different cell lines, which aids in preliminary identification.
Table 1: CPE Patterns and Identification of Common Viruses in Cell Culture
| Virus | Fibroblasts | A549 Cells | RhMK Cells | Final Identification |
|---|---|---|---|---|
| Adenovirus | Some produce clusters | Grape-like clusters or "lacy" pattern; 5–8 days | Some produce clusters | IF for group and neutralization for type [5] |
| Cytomegalovirus | Foci of contiguous rounded cells; 10–30 days | — | — | CPE [5] |
| Herpes Simplex Virus | Rounded large cells; 2–6 days | Rounded large cells; 1–4 days | Some produce CPE | IF for group and neutralization for type [5] |
| Influenza Virus | — | — | Undifferentiated CPE, cellular granulation; 4–8 days | IF for group and neutralization for type [5] |
| Rhinovirus | Degeneration, rounding; 7–10 days | — | — | CPE [5] |
Figure 1: Workflow for traditional tube cell culture virus isolation, highlighting the lengthy observation period for CPE development.
Modern Cell Culture Formats: Technological Advances
Modern cell culture formats have revolutionized virology by addressing key limitations of traditional methods, particularly regarding turnaround time, sensitivity, and scope of detection. These advances include novel culture vessels, cryopreservation techniques, co-culture systems, and engineered cell lines.
The shell vial or 1-dram vial has largely replaced the traditional screw-cap tube as the standard container. This smaller format allows for monolayer growth on the vial bottom and facilitates centrifugation-enhanced inoculation [5] [89]. Microwell plates (24- or 96-well formats, also called cluster plates) represent another significant advancement, enabling higher throughput testing [5] [89]. A critical innovation is the centrifuge-enhanced assay, where low-speed centrifugation of the shell vial post-inoculation rapidly forces viral particles into contact with host cells, drastically reducing the incubation time required for infection establishment [89].
Advanced Modern Techniques
- Cryopreserved Cell Culture: Monolayer cells are grown in shell vials and cryopreserved at -196°C in liquid nitrogen. Prior to use, vials are thawed in a 37°C water bath and prepared according to standardized protocols. This method maintains cell sensitivity to pathogens like Chlamydiae, CMV, and HSV while simplifying cell line maintenance and availability [5].
- Virus Isolation in Co-cultured Cells: This approach combines different cell types in a single vial to create a broader susceptible platform for virus isolation. For example:
- R-Mix Cells: A combination of A549 and mink lung cells in a shell vial is used for isolating various respiratory pathogens. After 24 hours of incubation, cells are stained with a fluorescein-labeled monoclonal antibody cocktail against adenoviruses, parainfluenza viruses (Types 1, 2, 3), influenza viruses (Types A and B), and respiratory syncytial virus [5].
- MRC-5 and A549 Coculture: Used for simultaneous detection of CMV, HSV, and adenoviruses. A cocktail of primary antibodies is applied, followed by secondary antibodies labeled with distinct fluorophores (e.g., FITC, Cy3). This multiplexing allows for detection of multiple viruses from a single sample inoculation [5].
- Virus Identification in Transgenic Cell Lines: Genetically engineered cell lines contain reporter genes that activate upon infection with a specific virus. For instance, a CD4-positive lymphoid cell line transformed with a retroviral vector containing a long terminal repeat promoter coupled to a chloramphenicol acetyltransferase (CAT) reporter gene can be used for HIV detection. Viral protein production activates the reporter, allowing detection before full virion assembly, thus reducing turnaround time [5] [55].
- Cell Combos Micromethod: A recently developed (2025) approach inoculates combinations of cell lines (e.g., Caco-2/MRC-5 combo) in a micro-method format to isolate a broad panel of respiratory viruses, including those potentially missed by molecular techniques like multiplex PCR. This strategy is particularly valuable for detecting unexpected or emerging viruses during unexplained outbreaks [19].
- Three-Dimensional (3D) Cell Culture Models: Emerging 3D technologies, such as air-liquid interface (ALI) cultures of human airway epithelial cells and stem cell-derived alveolar type II (AT2) cells, offer a more physiologically relevant model. These systems better simulate the in vivo microenvironment and have proven successful in isolating contemporary seasonal human coronaviruses (sHCoVs) that are notoriously difficult to grow in standard immortalized cell lines [3] [91].
Detailed Protocol: Shell Vial Culture with Immunofluorescence Staining (Pre-CPE Detection)
- Cell Preparation and Inoculation: Seed susceptible cells (e.g., MRC-5, A549, or co-cultured R-Mix) onto a coverslip within a shell vial and grow to confluence. Inoculate the vial with 0.2 mL of processed clinical sample [5] [89].
- Centrifugation and Incubation: Centrifuge the inoculated vial at 700 x g for 45-60 minutes at room temperature to enhance viral adsorption. Add maintenance medium and incubate at 35°C–37°C with 5% CO₂ for 24-48 hours. The incubation period is virus-dependent but is significantly shorter than traditional culture [5] [89].
- Staining and Detection: Remove the culture medium and fix the cell monolayer on the coverslip with acetone or methanol. Add virus-specific fluorescein-labeled monoclonal antibodies. Incubate, then wash to remove unbound antibody. Examine the coverslip using a fluorescence microscope for specific apple-green fluorescence, indicating a positive infection [5] [89].
Figure 2: Workflow for modern shell vial culture with pre-CPE detection, demonstrating the significantly reduced time-to-result.
Comparative Analysis: Performance and Applications
The transition from traditional to modern cell culture formats has yielded substantial improvements in diagnostic efficiency, scope, and application. The table below provides a direct comparison of key performance metrics.
Table 2: Comprehensive Comparison of Traditional and Modern Cell Culture Formats
| Parameter | Traditional Tube Culture | Modern Formats (Shell Vial, Co-culture, etc.) |
|---|---|---|
| Primary Format | Screw-cap tube (16 mm x 125 mm) [5] | Shell vial, microwell plate (24-/96-well) [5] |
| Time to Result | 5–10 days to several weeks [5] [1] | 24–48 hours for many viruses [5] [1] [89] |
| Detection Principle | Observation of Cytopathic Effect (CPE) [5] | Pre-CPE detection via immunofluorescence, reporter genes [5] [89] [55] |
| Throughput | Low | Medium to High (especially cluster plates) [5] |
| Sensitivity | High (Gold Standard) [5] | Enhanced sensitivity for many viruses [5] [1] |
| Multiplexing Capability | Low (requires multiple cell line tubes) | High (co-cultured cells, antibody cocktails) [5] [19] |
| Cost per Test | $1.5 - $6.50/tube [5] | Varies; can be lower due to reduced labor and time |
| Key Advantage | Gold standard, provides viable virus for further study [5] [55] | Speed, sensitivity, ability to detect fastidious viruses [5] [19] [91] |
| Key Limitation | Slow turnaround, labor-intensive, requires multiple cell lines [5] | May require specific reagents/antibodies, limited by design |
Integration with Molecular Methods
Modern cell culture often integrates with molecular techniques to overcome its own limitations and to provide a more comprehensive diagnostic picture. For instance, contemporary sHCoVs isolated in ALI cultures are confirmed and characterized using quantitative RT-PCR and next-generation sequencing [91]. Furthermore, molecular markers are being developed to surrogate viral culture. For SARS-CoV-2, detection of subgenomic RNA (sgRNA), particularly from the envelope (E) gene, has shown high accuracy (98%) in identifying viable virus compared to cell culture, offering a practical tool for clinical management [88].
The Scientist's Toolkit: Essential Research Reagent Solutions
The table below lists key reagents, cell lines, and materials essential for implementing the cell culture methods discussed in this analysis.
Table 3: Key Research Reagent Solutions for Virus Isolation Culture
| Reagent/Material | Function/Application | Examples & Notes |
|---|---|---|
| Primary & Diploid Cell Lines | Susceptible substrates for a wide range of viruses; used in both traditional and modern formats. | RhMK cells (influenza, parainfluenza); MRC-5 cells (CMV, VZV, rhinovirus); Human Foreskin Fibroblasts (HSV, CMV) [5]. |
| Continuous/Immortalized Cell Lines | Easy to maintain, used for virus propagation and in co-culture systems. | A549 (respiratory viruses); HEp-2 (respiratory syncytial virus, Mycoplasma pneumoniae [92]); Vero E6 (SARS-CoV-2 [88]) |
| Co-cultured Cell Systems | Broad-spectrum virus isolation in a single vial, increasing diagnostic efficiency. | R-Mix Cells (A549 + Mink Lung cells for respiratory viruses [5]); MRC-5/A549 combo [5] |
| Specialized Culture Models | Isolation of fastidious viruses and physiologically relevant studies. | Air-Liquid Interface (ALI) cultures (differentiated primary HNECs, BCi for sHCoVs [91]); Stem cell-derived AT2 cells [91] |
| Virus-Specific Monoclonal Antibodies | Essential for pre-CPE identification and confirmation in modern assays (IF). | Antibodies against influenza A/B, RSV, adenovirus, parainfluenza, HSV, CMV [5] [89] |
| Cryopreservation Media | Long-term storage of cell stocks and ready-to-use cryopreserved cell vials. | Typically contains culture medium, serum (e.g., FBS), and a cryoprotectant like DMSO or glycerol [5] |
| Shell Vials & Cluster Plates | Core physical platforms for modern, rapid cell culture. | 1-dram shell vials with coverslips; 24-well or 96-well plates [5] [89] |
The comparative analysis between traditional and modern cell culture formats reveals a clear trajectory toward faster, more sensitive, and more physiologically relevant systems. Traditional tube culture, while remaining the gold standard for virus isolation and providing viable isolates for further characterization, is hampered by its long turnaround time and labor-intensive nature. Modern formats, including shell vials, co-culture systems, transgenic cell lines, and advanced 3D models, have successfully addressed these limitations. They significantly reduce the time to diagnosis—from weeks to days or even hours—while improving detection sensitivity for a broad spectrum of viruses, including those that are non-cytopathic or difficult to culture.
The choice between traditional and modern methods is not always binary and depends on the specific research or diagnostic objectives. Traditional methods are indispensable for initial virus isolation, phenotypic characterization, and vaccine development. In contrast, modern methods are superior for rapid diagnostics, high-throughput screening, and studying viruses in models that closely mimic human physiology. The ongoing integration of cell culture with molecular techniques like sgRNA detection and next-generation sequencing creates a powerful synergistic toolkit for virology. Future prospects point toward the increased use of personalized 3D models and engineered reporter cell lines, further solidifying the role of cell culture as a fundamental and evolving technology in virus research and drug development.
The isolation of viruses using cell culture remains a cornerstone of virology, providing essential insights into viral pathogenicity, host interactions, and therapeutic development [6]. Traditional cell culture methods alone, however, face significant challenges including difficulty in detecting non-cytopathic viruses and the persistent risk of viral contamination in cell lines [6]. The integration of molecular methods—specifically polymerase chain reaction (PCR) and whole genome sequencing (WGS)—with classical cell culture techniques creates a powerful synergistic framework that enhances the speed, sensitivity, and analytical depth of virological research. This integrated approach is particularly valuable for early pathogen detection in public health surveillance [93], characterization of emerging viral variants [93], and ensuring the safety of biological products [6]. This protocol details the systematic integration of these methodologies to optimize virus isolation and characterization within a research context.
Experimental Protocols and Workflows
Integrated Workflow for Virus Isolation and Characterization
The following diagram illustrates the comprehensive process for isolating viruses from environmental or clinical samples and subsequent molecular characterization.
Figure 1: Integrated workflow for virus isolation and molecular characterization.
Sample Processing and Viral Concentration
2.2.1 Collection and Pre-processing
- Sample Types: Collect wastewater (24-hour flow-weighted composite or grab samples) or clinical specimens (e.g., respiratory secretions, stool samples) [93].
- Transport and Storage: Maintain samples at 4°C during transport and process within 2-4 hours of collection. Avoid freezing, as it significantly reduces RNA yield [94].
- Debris Removal: Centrifuge samples at 4,000 × g for 30 minutes at 4°C to remove large particulates [93] [94].
2.2.2 Viral Concentration Methods
- PEG Precipitation: Incubate supernatant with 8% PEG-8000 and 0.3 M NaCl overnight (~16 hours) at 4°C. Centrifuge at 10,000 × g for 120 minutes at 4°C. Resuspend pellet in 500 μL Opti-MEM or molecular biology grade water [93] [94].
- Centrifugal Filtration: Using Vivaspin devices (MWCO 50 kDa), concentrate supernatant by centrifuging at 4,000 × g for 30 minutes at 4°C. Repeat until entire sample volume is processed and final concentrate reaches 500 μL [94].
- Alternative Kits: Commercial kits like Zymo Environ Water RNA Kit combine viral enrichment and RNA purification in one workflow, providing superior RNA quality compared to PEG precipitation and ultrafiltration methods [94].
Cell Culture Propagation for Virus Isolation
2.3.1 Cell Line Selection and Maintenance
- HEp-2 Cells: Maintain in complete DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C with 5% CO₂. For virus culture, use DMEM with 2% FBS and antibiotics [85].
- H1HeLa Cells: Preferred for rhinovirus propagation. Ideal for high-titer virus production required for host response studies [86].
- A549 Cells: Suitable for studying cytopathic effects of various respiratory viruses including adenovirus and HSV-2 [6].
2.3.2 Virus Inoculation and Passage
- Inoculate cell monolayers with filtered (0.22 μm) clinical samples or concentrated environmental samples [85].
- Incubate at 37°C for up to 7 days, monitoring daily for cytopathic effects (CPE) including cell rounding, syncytia formation, and cell lysis [85] [6].
- If no CPE is observed after 7 days, use culture supernatants to inoculate fresh cells for 2 additional passages. For adenovirus-like CPE, passage again to confirm viral presence [85].
- Collect virus-infected cells and supernatant for subsequent detection and genome sequencing [85].
Molecular Detection and Characterization
2.4.1 Nucleic Acid Extraction
- Extract viral RNA using column-based kits (e.g., NucleoSpin RNA Virus, Promega Viral RNA/DNA Extraction Kit, or Zymo Environ Water RNA Kit) according to manufacturer's protocols [93] [94].
- Elute RNA in 30-100 μL TE buffer or RNase-free water and store at -80°C for downstream analyses [93].
- Include internal positive controls (IPC) prior to RNA extraction to calculate recovery efficiency and assess RT-qPCR inhibition [93].
2.4.2 Detection Methods Comparison
Table 1: Comparison of SARS-CoV-2 Detection Methods in Complex Samples
| Method | Target Genes | Limit of Detection | Advantages | Disadvantages |
|---|---|---|---|---|
| RT-qPCR | N gene, RdRP, E | 2.13 copies/reaction (95% CI) [94] | High throughput, standardized protocols | Susceptible to inhibition, semi-quantitative |
| RT-ddPCR | RdRP, E | Improved sensitivity for low viral load [94] | Absolute quantification, resistant to inhibition | Higher cost, specialized equipment required |
| Multiplex Panels | Multiple respiratory pathogens | Varies by panel | Comprehensive pathogen screening | May have lower sensitivity for individual targets |
2.4.3 PCR Protocols
- RT-qPCR: Perform one-step reactions using 5 μL extracted RNA in 20 μL total volume. Conditions: reverse transcription at 42°C for 30 min; initial denaturation at 95°C for 3 min; 50 cycles of 95°C for 10 s and 60°C for 30 s [94].
- RT-ddPCR: Prepare 20 μL reactions with 3 μL sample, 5 μL supermix, 2 μL reverse transcriptase, and 1 μL 300 mM DTT. Partition samples into droplets and run with same thermal cycling conditions as RT-qPCR [94].
2.4.4 Whole Genome Sequencing
- Use Illumina platforms (e.g., MiSeq with COVIDSeq Assay Index kit and MiSeq Reagent Kit V3) [93].
- Library preparation: Reverse transcribe SARS-CoV-2 RNA to cDNA, then amplify viral genome using ARTIC v4 primer cocktails generating 99 amplicons [93].
- Process raw sequencing data through quality control trimming, reference mapping assembly, and lineage prediction using HAVoC pipeline or Pangolin [93].
Research Reagent Solutions
Table 2: Essential Research Reagents for Integrated Virology Workflows
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cell Culture Media | DMEM with 10% FBS, antibiotics | Cell maintenance and propagation [85] |
| Virus Culture Media | DMEM with 2% FBS | Supports viral replication with reduced cellular metabolism [85] |
| Nucleic Acid Extraction Kits | Zymo Environ Water RNA Kit, NucleoSpin RNA Virus, Promega Viral RNA/DNA Kit | Viral RNA extraction from complex samples [93] [94] |
| PCR Master Mixes | ViroReal Kit SARS-CoV-2, gb SARS-CoV-2 Multiplex, One-Step RT-ddPCR Advanced Kit | Detection and quantification of viral targets [93] [94] |
| Sequencing Kits | COVIDSeq Assay, MiSeq Reagent Kits | Library preparation and whole genome sequencing [93] |
| Concentration Reagents | PEG-8000, NaCl, Vivaspin centrifugal filters | Viral particle concentration from large volume samples [93] [94] |
Key Experimental Data and Applications
Method Performance and Validation
4.1.1 Longitudinal Monitoring Data A year-long wastewater monitoring campaign in Bucharest demonstrated that SARS-CoV-2 concentrations in wastewater preceded the increase in clinical cases by nearly 2 weeks, highlighting the predictive value of this integrated approach [93]. The study collected approximately 300 samples twice weekly from a wastewater treatment plant and an infectious diseases hospital, with higher raw concentrations observed in hospital samples, though urban monitoring provided more epidemiologically relevant data after population normalization [93].
4.1.2 Method Comparison Studies Comparative studies of viral concentration and detection methods revealed that:
- The Zymo Environ Water RNA Kit provided superior RNA quality compared to both PEG precipitation and ultrafiltration methods [94].
- Freezing wastewater samples significantly reduces RNA yield and should be avoided when possible [94].
- RT-ddPCR outperforms RT-qPCR in both specificity and sensitivity, particularly for samples with low viral load [94].
Table 3: Viral Pathogens Detectable via Integrated Culture-Molecular Approaches
| Virus Category | Specific Pathogens | Recommended Cell Lines | CPE Observations |
|---|---|---|---|
| Respiratory Viruses | SARS-CoV-2, Rhinovirus A/B | H1HeLa, A549, Vero E6 | Cell rounding, detachment [6] [86] |
| Herpesviruses | Epstein Barr Virus (EBV), Ovine Herpesvirus 2 (OvHV-2) | Primary B-cells, various mammalian lines | Lymphoblastoid transformation, cell lysis [6] |
| Gastrointestinal Viruses | Adenovirus, Bocavirus, Reovirus | HEp-2, A549 | Cell aggregation, granulation [93] [6] |
Advanced Applications and Troubleshooting
4.2.1 Viral Viability Assessment While molecular methods detect viral genetic material, cell culture remains essential for determining infectivity. In wastewater studies, only a few SARS-CoV-2 isolates could demonstrate persistence during in vitro passages, highlighting the importance of culture for validating viral viability despite lower success rates [93].
4.2.2 Contamination Control Viral contamination in cell culture poses significant challenges, particularly with ubiquitous viruses like Epstein Barr Virus (EBV) which infects approximately 98% of humans, and Ovine Herpesvirus 2 (OvHV-2) which can infect numerous species [6]. Regular screening using PCR assays and implementing robust quality control measures including STR profiling and mycoplasma testing are essential for maintaining culture integrity [6].
4.2.3 Multiplex Pathogen Surveillance Integrated culture-PCR approaches enable comprehensive pathogen monitoring. In wastewater surveillance, adenovirus, bocavirus and reovirus were identified as the most abundant viruses in both urban and hospital wastewater, demonstrating the utility of this approach for tracking multiple pathogens simultaneously [93].
The integration of molecular methods with cell culture isolation creates a powerful framework for advanced virology research. This synergistic approach leverages the sensitivity of PCR and sequencing with the biological relevance of cell culture, enabling more comprehensive virus characterization, earlier detection of emerging pathogens, and more accurate assessment of infectivity. As viral threats continue to evolve, these integrated protocols provide researchers with robust tools for public health surveillance, drug development, and fundamental virological investigation. The standardized methodologies presented here offer a reproducible template for implementing this integrated approach across diverse research and public health settings.
Validation Through Immunofluorescence, Hemagglutination, and Nucleic Acid Tests
Within virus isolation research, the reliability of cell culture methods is paramount. Validating the presence, identity, and effects of a viral isolate requires a suite of highly specific and sensitive detection techniques. This document provides detailed application notes and protocols for three cornerstone methods: Immunofluorescence, which allows for the spatial localization of viral antigens within cultured cells; the Hemagglutination Inhibition (HI) Assay, a classic method for detecting and quantifying specific anti-viral antibodies; and Nucleic Acid Testing, which identifies viral genetic material. Proper validation of these assays is critical for confirming viral isolation, characterizing immune responses, and ensuring the safety of biopharmaceutical products derived from cell culture systems.
The following tables summarize key performance characteristics and parameters for the validation of the three analytical techniques, providing a clear framework for assay evaluation.
Table 1: Key Analytical Performance Characteristics for Assay Validation
| Performance Characteristic | Immunofluorescence (TUNEL/MILAN) [95] | Hemagglutination Inhibition (HI) [96] [97] [98] | Nucleic Acid Testing (Multiplex) [99] [100] |
|---|---|---|---|
| Selectivity/Specificity | Specific for DNA fragmentation in cell death; compatible with protein antigen colocalization. | Highly selective, allowing clear discrimination between positive and negative serum samples [96]. | Ability to detect intended targets without cross-reactivity in a multiplex format [100]. |
| Sensitivity | Detects individual apoptotic or necrotic cells in situ. | Reliable detection at a starting serum dilution of 1:10 [97]. | Determined by the Limit of Detection (LOD) for each target [99]. |
| Precision (Repeatability) | Consistent TUNEL signal across antigen retrieval methods [95]. | High intra-assay, inter-assay, and total assay precision (%GCV) [97]. | Consistent results across multiple test runs [100]. |
| Accuracy | Qualitatively matches commercial TUNEL kit results [95]. | Accurate as evidenced by low % bias measurements [97]. | Agreement with a reference method or known samples. |
| Linearity & Range | Not typically quantified for this imaging method. | Linear correlation between low and high antibody levels [96] [98]. | Quantitative linear range for each target [100]. |
| Robustness | Robust to changes in antigen retrieval method (Pressure Cooker vs. Proteinase K) [95]. | Consistent results when serum-antigen interaction times are altered [96]. | Consistent performance under small, deliberate changes in protocol parameters. |
Table 2: Summary of Critical Experimental Parameters
| Parameter | Immunofluorescence (TUNEL) [95] | Hemagglutination Inhibition (HI) [96] [97] | Nucleic Acid Testing [99] [100] |
|---|---|---|---|
| Key Reagent | Terminal deoxynucleotidyl transferase (TdT) | Red Blood Cells (RBCs; chicken, human type O) | Primers/Probes for target sequences |
| Sample Type | Formalin-Fixed Paraffin-Embedded (FFPE) tissue sections | Human or animal serum | Cell culture supernatant, extracted nucleic acids |
| Critical Step | Antigen retrieval (Pressure cooker preferred over Proteinase K) | Removal of non-specific serum inhibitors with RDE | Nucleic acid extraction and amplification |
| Assay Readout | Fluorescence microscopy | Hemagglutination pattern in microtiter plates | Fluorescence from DNA binding dyes or probes |
| Validation Focus | Compatibility with multiplexed protein detection (e.g., MILAN) | Precision, specificity, and reproducibility of antibody titer | Limit of detection, specificity, and inclusivity |
Detailed Experimental Protocols
Protocol: Harmonized TUNEL and Multiplexed Immunofluorescence (MILAN) for Cell Death Validation
This protocol harmonizes the TUNEL assay with Multiple Iterative Labeling by Antibody Neodeposition (MILAN), enabling the spatial contextualization of cell death within a rich proteomic landscape of virus-infected cell cultures [95].
I. Materials
- Terminal deoxynucleotidyl transferase (TdT) and reaction buffer
- Fluorophore-conjugated dUTP (e.g., BrdU, EdU)
- Antibodies: Primary antibodies against viral antigens/cell markers, Anti-BrdU (if using BrdU), and species-specific secondary antibodies with different fluorophores.
- Pressure Cooker for antigen retrieval
- Erasure Buffer: 2-Mercaptoethanol (2-ME) with Sodium Dodecyl Sulfate (SDS)
- Blocking Buffer: Proteinase K is not recommended as it degrades protein antigenicity.
II. Methodology
- Sample Preparation: Culture cells on coverslips or prepare FFPE cell pellets. Infect with virus and fix at appropriate timepoints.
- Antigen Retrieval: Perform heat-induced epitope retrieval using a pressure cooker. Do not use Proteinase K, as it compromises subsequent protein immunofluorescence [95].
- TUNEL Reaction: a. Prepare TUNEL reaction mixture per manufacturer's instructions (TdT enzyme, reaction buffer, and fluorophore-labeled dUTP). b. Apply mixture to samples and incubate in a humidified chamber at 37°C for 60-90 min. c. Wash thoroughly to terminate the reaction.
- Immunofluorescence Staining (Cycle 1): a. Block samples with an appropriate blocking buffer. b. Apply primary antibody (e.g., against a viral protein). c. Apply fluorophore-conjugated secondary antibody. d. Image the sample to capture TUNEL and first-round protein signals.
- Antibody Erasure: a. De-coverslip (if mounted) and incubate slides in 2-ME/SDS erasure buffer at 66°C to remove primary and secondary antibodies [95]. b. Wash thoroughly.
- Iterative Staining (Cycle 2-n): a. Repeat Step 4 with a new set of primary and secondary antibodies for different protein targets. b. After imaging, repeat Step 5 for erasure. c. Continue for subsequent cycles to build a multiplexed protein dataset.
III. Workflow Diagram
Protocol: Validated Hemagglutination Inhibition (HI) Assay for Anti-Viral Antibody Detection
This protocol describes a validated HI assay suitable for quantifying antibodies in serum from subjects vaccinated with viral vectors or infected with hemagglutinating viruses [96] [97] [98].
I. Materials
- Viral Antigen: Inactivated virus or recombinant Virus-Like Particles (VLPs) with known hemagglutinating units (HU) [97].
- Red Blood Cells (RBCs): 0.75% suspension of human Type O or chicken RBCs in DPBS [96] [97].
- Test Sera: Human or animal serum samples.
- Receptor-Destroying Enzyme (RDE): From Vibrio cholerae filtrate.
- Microtiter Plates: 96-well U-bottom plates.
II. Methodology
- Serum Pre-treatment: a. Dilute serum 1:4 with RDE. b. Incubate at 37°C for 18-20 hours to destroy non-specific inhibitors. c. Heat-inactivate at 56°C for 30 minutes. d. Dilute to a final 1:10 concentration in DPBS [97].
- Viral Antigen Standardization: a. Confirm the titer of the viral antigen (HU) by serial dilution in DPBS. b. Use the concentration equivalent to 8 HU per 50 µL for the assay.
- HI Assay Procedure: a. Add 25 µL of DPBS to all wells of a U-bottom plate. b. Add 25 µL of pre-treated serum to the first well. Serially dilute 2-fold across the plate. c. Add 25 µL of antigen (8 HU) to each well. Include serum and cell controls. d. Shake the plate gently and incubate at room temperature for a defined period (e.g., 30-60 min) [96]. e. Add 50 µL of 0.75% RBC suspension to each well. f. Incubate at 4°C until a distinct button forms in the negative control well (approximately 60-90 min).
- Reading and Interpretation: a. The HI titer is the reciprocal of the highest serum dilution that completely inhibits hemagglutination, forming a distinct button or a dot that runs in a tear-drop shape when the plate is tilted [96] [97]. b. Automated readers can be used for objective endpoint determination [97].
III. Workflow Diagram
Protocol: Validation of Nucleic Acid Tests for Viral Detection
This protocol outlines the key steps for validating nucleic acid amplification tests, including multiplex assays, for the detection of viral pathogens in cell culture samples, in alignment with clinical and biopharmaceutical guidelines [99] [100].
I. Materials
- Nucleic Acid Extraction Kit: Validated for the sample type.
- Primers and Probes: Specific for the target viral sequence(s).
- Internal Control: To monitor extraction and amplification efficiency.
- Master Mix: Contains polymerase, dNTPs, and buffer.
- Real-Time PCR Instrument.
II. Methodology
- Validation Experiment Design: a. Limit of Detection (LOD): Determine the lowest concentration of viral target that can be detected ≥95% of the time using a dilution series of standardized material [99]. b. Specificity/Inclusivity: Test the assay against a panel of related viral strains (to ensure detection) and other unrelated pathogens or human nucleic acids (to ensure no cross-reactivity) [100]. c. Linearity and Range: Assess the quantitative linear range by testing known concentrations of the target and evaluating the correlation between the input and the quantitative result (e.g., Ct value). d. Precision: Perform repeatability (within-run) and reproducibility (between-run, between-operator, between-instrument) studies.
- Routine Testing Procedure: a. Extract nucleic acids from cell culture supernatant or lysates, including an internal control. b. Prepare the PCR reaction mix with master mix, primers/probes, and template. c. Run the amplification protocol on a real-time PCR instrument. d. Analyze results based on the established validation parameters (e.g., Ct value below a predetermined cutoff indicates a positive result).
III. Workflow Diagram
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Validation Assays in Virus Research
| Reagent / Solution | Function / Application | Key Consideration / Example |
|---|---|---|
| Receptor-Destroying Enzyme (RDE) | Removes non-specific hemagglutination inhibitors from mammalian serum samples prior to HI testing [96] [97]. | Derived from Vibrio cholerae filtrate; requires long incubation (18-20 hrs) [97]. |
| Virus-Like Particles (VLPs) | Non-infectious agglutinins for HAI assays; present native viral surface proteins without risk of infection [97]. | Allow use of wild-type sequences, avoiding serological bias from egg-adapted mutations [97]. |
| Terminal Deoxynucleotidyl Transferase (TdT) | Core enzyme in TUNEL assay; catalyzes the addition of labeled dUTP to free 3'-OH ends of fragmented DNA [95]. | Labels DNA breaks characteristic of apoptotic and necrotic cell death for fluorescence detection. |
| 2-Mercaptoethanol/SDS Erasure Buffer | Key component for multiplexed IF (MILAN); removes antibodies from FFPE sections between staining cycles [95]. | Enables iterative staining (>20 cycles) on a single sample, preserving tissue integrity [95]. |
| Accessible Color Palettes | For creating clear, interpretable diagrams and figures that are legible to all audiences, including those with color vision deficiencies [101]. | Follow WCAG guidelines; ensure high contrast (≥4.5:1); avoid problematic combinations like red/green [101]. |
| Multiplex Nucleic Acid Controls | Validated reference materials used to ensure that a multiplex NAT correctly identifies all intended targets [100]. | Essential for establishing assay specificity, LOD, and preventing false positives/negatives during validation [100]. |
Assessing Sensitivity, Specificity, and Cost-Effectiveness of Different Approaches
Virus detection and isolation are critical for diagnosing infections, studying viral ecology, and developing therapeutics. This application note provides a comparative assessment of two advanced methodological approaches: a novel double-stranded RNA (dsRNA) extraction method for high-throughput sequencing (HTS) virome profiling and a cell culture-based micromethod for isolating respiratory viruses. We evaluate their sensitivity, specificity, and cost-effectiveness, providing detailed protocols and resource guides to facilitate implementation in virology and drug development research.
Effective viral disease management hinges on the ability to accurately monitor viruses and anticipate outbreaks. Cell culture remains a cornerstone technique for virus isolation, particularly for detecting unknown or emerging pathogens that may evade molecular detection [19]. Concurrently, high-throughput sequencing (HTS) offers powerful, unbiased detection of viral communities. This document frames the comparison of a modernized cell culture technique and a novel sequencing-based extraction method within the broader research on cell culture methods for virus isolation, providing a framework for selecting context-appropriate diagnostic strategies.
Comparative Analysis of Virus Detection Methods
The table below summarizes the key performance metrics and characteristics of the dsRNA extraction and cell culture combo methods, based on recent studies.
Table 1: Comparison of Virus Detection Method Performance and Characteristics
| Feature | B2-Based dsRNA Extraction for HTS [102] | Cell Combos Micromethod for Virus Isolation [19] |
|---|---|---|
| Core Principle | Protein-based purification of viral dsRNA for sequencing | Inoculation of clinical samples onto permissive cell line combinations to enable viral growth |
| Detection Specificity | 0.97 (minimizes false positives) [102] | High (confirmed by cytopathic effect and PCR) [19] |
| Detection Sensitivity | 0.71 [102] | Effective for viruses not detected by multiplex RT-PCR; isolated 12 viruses from PCR-negative samples in proof-of-concept [19] |
| Cost-Effectiveness | Highly cost-effective at $4.47 per reaction [102] | Cost not explicitly stated, but micromethod design reduces sample volume requirements, conserving reagents [19] |
| Key Advantage | Excellent for virome profiling and ecology studies; high viral read purity [102] | Capable of detecting unexpected, genetically divergent, or emerging viruses [19] |
| Primary Application | Viral discovery, virome-host interactions, and ecology [102] | Diagnostic investigation of undiagnosed respiratory outbreaks and virus isolation [19] |
Experimental Protocols
Protocol: B2-Based dsRNA Extraction for High-Throughput Sequencing
This protocol describes a bead-free and resin-free method for extracting dsRNA using the Flock House virus B2 protein, which binds dsRNA electrostatically [102].
Key Materials:
- Flock House virus (FHV) B2 protein
- Lysis/Binding Buffer
- Wash Buffer
- Elution Buffer
- Nuclease-free water and tubes
Procedure:
- Sample Lysis: Homogenize the cell culture or tissue sample in an appropriate Lysis/Binding Buffer.
- dsRNA Binding: Incubate the lysate with the B2 protein. The protein will electrostatically bind to dsRNA in the sample.
- Washing: Separate the B2-dsRNA complexes from the lysate and wash with a suitable Wash Buffer to remove contaminants like single-stranded RNA and proteins.
- Elution: Elute the purified dsRNA using an Elution Buffer.
- Downstream Application: The eluted dsRNA is now ready for library preparation and HTS.
Performance Notes: This method yielded viral read proportions exceeding 20% in most samples, with less co-extraction of low-weight molecules compared to cellulose-based and DRB4-based methods [102].
Protocol: Cell Combos Micromethod for Isolating Respiratory Viruses
This protocol details the use of combinations of cell lines in a micromethod format to isolate a broad panel of respiratory viruses, including those missed by molecular techniques [19].
Key Materials:
- Cell Lines: Ten selected cell lines, combined into five two-line "combos." The Caco-2/MRC-5 combo was identified as particularly promising [19].
- Media: Appropriate growth media for the selected cell lines.
- Clinical Samples: Respiratory samples (e.g., nasopharyngeal swabs).
Procedure:
- Cell Culture Preparation: Grow the pre-selected cell line combinations (e.g., Caco-2/MRC-5) in culture plates or flasks.
- Sample Inoculation: Inoculate the cell combos with a small volume of the clinical sample. The micromethod requires less sample volume than standard culture or metagenomics.
- Incubation and Monitoring: Incubate the cultures and monitor daily for the appearance of a cytopathic effect (CPE).
- Virus Detection: Confirm viral multiplication by observing CPE and/or through follow-up RT-PCR assays.
Performance Notes: In a proof-of-concept study using 859 multiplex RT-PCR-negative respiratory samples, this approach successfully isolated 12 herpes simplex or varicella-zoster viruses that were not initially detected [19].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Viral Detection and Isolation Protocols
| Item | Function/Application |
|---|---|
| Flock House Virus (FHV) B2 Protein | Core reagent for the novel, cost-effective dsRNA extraction method; binds dsRNA electrostatically [102]. |
| Caco-2 Cell Line | A human epithelial colorectal adenocarcinoma cell line; part of the most promising cell combo for isolating a broad range of respiratory viruses [19]. |
| MRC-5 Cell Line | A human fetal lung fibroblast cell line; used in combination with Caco-2 for enhanced virus isolation [19]. |
| C6/36 Cell Line | Derived from Aedes albopictus mosquito larvae; commonly used for the amplification of arboviruses like Dengue virus [103]. |
| BHK-21 Cell Line | Baby Hamster Kidney fibroblast cell line; used in viral amplification and plaque assays [103]. |
| Amicon Ultra 100 kDa Centrifugal Filters | Used for concentrating viral stocks, increasing titer, and improving long-term stability [103]. |
| Carboxymethylcellulose | A viscous medium used in overlay solutions for plaque assays to restrict viral diffusion and enable plaque formation [103]. |
Workflow and Pathway Visualizations
The following diagrams illustrate the logical workflows for the two primary methods discussed.
Diagram 1: Workflow for B2-based dsRNA extraction and sequencing. This process leverages the electrostatic properties of the B2 protein for bead-free purification of viral dsRNA [102].
Diagram 2: Workflow for cell combo micromethod virus isolation. This updated culture approach uses minimal sample volume to detect known and emerging viruses [19].
Conclusion
Cell culture remains an indispensable tool for virus isolation, continuously evolving through methodological innovations while maintaining its status as the gold standard for viral diagnostics and research. The integration of traditional techniques with modern approaches like cryopreservation, co-cultured systems, and molecular validation has significantly enhanced efficiency, specificity, and application scope. Future directions point toward increased automation, further refinement of 3D culture systems, and deeper integration with omics technologies, promising to expand capabilities in vaccine development, antiviral drug screening, and emerging pathogen response. For researchers and drug development professionals, mastering both foundational principles and advanced applications of viral cell culture will continue to be crucial for advancing biomedical science and therapeutic development.
References
- 1. Traditional and Modern Cell Culture in Virus Diagnosis [https://ophrp.org/journal/view.php?number=415]
- 2. What is cell culture, and how has it evolved? [https://virologyresearchservices.com/2024/07/07/what-is-cell-culture-and-how-has-it-evolved/]
- 3. Three-dimensional cell culture models in respiratory virus ... [https://pubs.rsc.org/en/content/articlelanding/2025/tb/d5tb00290g]
- 4. Halfway between 2D and Animal Models: Are 3D Cultures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5796130/]
- 5. Traditional and Modern Cell Culture in Virus Diagnosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4850366/]
- 6. Viral contamination in cell culture: analyzing the impact of ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1442321/full]
- 7. 3D Cell Cultures are Better than Animal Models - Here´s Why! [https://focus.gbo.com/blog/3d/advantages-of-3d-cell-culture-over-animal-models-greiner-bio-one]
- 8. A quantitative meta-analysis comparing cell models in ... [https://www.nature.com/articles/s41598-023-35043-5]
- 9. A Review of the Utility of Established Cell Lines for Isolation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11768479/]
- 10. Re-imagining human cell culture media: Challenges, ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025000503]
- 11. Isolation in Cell Culture - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7152012/]
- 12. 6.3: Isolation, Culture, and Identification of Viruses [https://bio.libretexts.org/Bookshelves/Microbiology/Microbiology_(OpenStax)/06%3A_Acellular_Pathogens/6.03%3A_Isolation_Culture_and_Identification_of_Viruses]
- 13. Virology Culture Guide [https://www.atcc.org/resources/culture-guides/virology-culture-guide]
- 14. Novel Protocol for the Preparation of Porcine Bone Marrow ... [https://www.mdpi.com/2409-9279/6/5/73]
- 15. Virus Isolation - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/virus-isolation]
- 16. Optimized Protocol for Isolation and Culture of Primary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12553468/]
- 17. Virology Lab Equipment [https://www.onepointesolutions.com/blog/virology-lab-equipment/]
- 18. Protocol for detection of Oropouche viruses from human ... [https://www.sciencedirect.com/science/article/pii/S2666166725002114]
- 19. Development of cell combos micromethod to isolate ... [https://pubmed.ncbi.nlm.nih.gov/41217198/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=0GaYCevddEDAfC5HA0wlElkFqaKqOUgRmbLrsDcYiaD&fc=None&ff=20251111123623&v=2.18.0.post22+67771e2]
- 20. How to Design a Virology Lab [https://www.onepointesolutions.com/blog/how-to-design-a-virology-lab/]
- 21. Safe Handling of Infectious Agents - Biosafety In The Laboratory [https://www.ncbi.nlm.nih.gov/books/NBK218635/]
- 22. Virology Laboratory Guidelines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8387051/]
- 23. Viral Isolation and Culture from Field-Collected Ticks: In Vitro [https://pubmed.ncbi.nlm.nih.gov/40512304/]
- 24. - Wikipedia Cytopathic effect [https://en.wikipedia.org/wiki/Cytopathic_effect]
- 25. of Viruses Protocols Cytopathic Effects [https://asm.org/protocols/cytopathic-effects-of-viruses-protocols]
- 26. Cytopathic Effect - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/cytopathic-effect]
- 27. : Types, Mechanisms & Examples Explained Cytopathic Effect [https://www.vedantu.com/biology/cytopathic-effect]
- 28. Cytopathic Effect Assay and Plaque Assay to Evaluate in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8855088/]
- 29. Measure viral-induced cytopathic effects with a quantitative ... [https://www.moleculardevices.com/en/assets/app-note/br/measure-viral-induced-cytopathic-effects-with-quantitative-luminescence-assay]
- 30. Enhanced isolation of respiratory syncytial virus in cell culture [https://pubmed.ncbi.nlm.nih.gov/3700634/]
- 31. CACO-2 cells: A continuous cell line with sensitive and ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093421000598]
- 32. Isolation of influenza virus in human lung embryonated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC228309/]
- 33. Alternative cell line for virus isolation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC268886/]
- 34. Comparison of HeLa-I, HEp-2 and NCI-H292 cell lines for ... [https://pubmed.ncbi.nlm.nih.gov/17825929/]
- 35. An engineered A549 cell line expressing CD13 and ... [https://www.sciencedirect.com/science/article/pii/S0042682223002088]
- 36. Comparison of standard tube and shell vial cell culture ... [https://pubmed.ncbi.nlm.nih.gov/2982911/]
- 37. Rapid Detection of Respiratory Viruses by Shell Vial Assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC85156/]
- 38. Isolation of seven respiratory viruses in shell vials [https://pmc.ncbi.nlm.nih.gov/articles/PMC262779/]
- 39. Microtiter Virus Isolation and Enzyme Immunoassays for ... [https://pubmed.ncbi.nlm.nih.gov/9157132/]
- 40. Development and Validation of a Microtiter Plate-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5923483/]
- 41. Comparison of Viral Isolation and Multiplex Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2650906/]
- 42. Identification of optimal sample collection devices and ... [https://pubmed.ncbi.nlm.nih.gov/32643291/]
- 43. Optimal specimen collection and transport methods for the ... [https://bmcvetres.biomedcentral.com/articles/10.1186/1746-6148-9-35]
- 44. Validating and optimizing the method for molecular ... [https://www.sciencedirect.com/science/article/pii/S0048969721065128]
- 45. An optimized nucleic acid isolation protocol for virus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8563463/]
- 46. Optimization of a more efficient protocol for mechanical ... [https://www.sciencedirect.com/science/article/abs/pii/S0261219417303599]
- 47. Laboratory Biosafety Guidelines for working with SARS- ... [https://www.cdc.gov/covid/php/lab/index.html]
- 48. Experimental Models to Investigate Viral and Cellular ... [https://www.mdpi.com/2076-2607/13/11/2444]
- 49. Technical pitfalls when collecting, cryopreserving, thawing ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1382192/full]
- 50. Phenotypic and functional comparisons between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576105/]
- 51. Tumor organoid-immune co-culture models: exploring a ... [https://www.nature.com/articles/s41420-025-02407-x]
- 52. A simple method for isolation of cell - associated particles from... viral [https://pubmed.ncbi.nlm.nih.gov/28923314/]
- 53. Comparison of the methods for isolation and detection ... [https://www.frontiersin.org/journals/public-health/articles/10.3389/fpubh.2023.1116636/full]
- 54. and Identification | SpringerLink Virus Isolation [https://link.springer.com/chapter/10.1007/978-1-4612-3900-0_3]
- 55. Cell Cultures for Virology: Usability, Advantages, and ... [https://www.mdpi.com/1422-0067/21/21/7978]
- 56. Virology through numbers: Plaque and TCID50 assays [https://virologyresearchservices.com/2024/07/15/virology-through-numbers-plaque-and-tcid50-assays/]
- 57. Comparison of the plaque assay and 50% tissue culture ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093413002024]
- 58. 50% Tissue Culture Infectious Dose (TCID50) for SARS- ... [https://www.protocols.io/view/50-tissue-culture-infectious-dose-tcid50-for-sars-df8g3rtw.html]
- 59. Development of an automated plaque-counting program ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11992114/]
- 60. Time to revisit the endpoint dilution assay and to replace ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8553042/]
- 61. A versatile automated pipeline for quantifying virus ... [https://www.nature.com/articles/s41467-024-49444-1]
- 62. Cell culture challenges: Contamination & prevention [https://greenelephantbiotech.com/blog/cell-culture-challenges-contamination-prevention/]
- 63. Viral contamination in cell culture: analyzing the impact of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11893573/]
- 64. A Beginner's Guide to Cell Culture: Practical Advice for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10000895/]
- 65. Protocols [https://unclineberger.org/tissueculture/protocols/]
- 66. Extracellular vesicles derived EBV tegument protein ... [https://www.nature.com/articles/s41467-025-64037-2]
- 67. Cell Culture Contamination: 5 Common Sources and How ... [https://www.capricorn-scientific.com/knowledge-center/cell-culture-contamination]
- 68. Comprehensive Microbial Control Strategy [https://www.bioprocessintl.com/qa-qc/beyond-traditional-microbiology-testing-developing-a-comprehensive-strategy-for-microbial-control-and-detection]
- 69. Troubleshooting Common Cell Culture Contamination Issues [https://cellculturecompany.com/troubleshooting-common-cell-culture-contamination-issues/]
- 70. Sensitive Detection of Cell Culture Mycoplasma Infection [https://www.rapidmicrobiology.com/news/sensitive-detection-of-cell-culture-mycoplasma-infection]
- 71. Innovative exploration of Hep-2 cell culture in the isolation ... [https://pubmed.ncbi.nlm.nih.gov/40370497/]
- 72. Novel method detects microbial contamination in cell cultures [https://news.mit.edu/2025/novel-method-detects-microbial-contamination-smart-0425]
- 73. Virus detection by short read high throughput sequencing ... [https://www.nature.com/articles/s41541-025-01104-1]
- 74. Comparison of two continuous cell lines BHK-21 and IB ... [https://pubmed.ncbi.nlm.nih.gov/40512422/]
- 75. Standardization of Quantitative Plaque-Based Viral Assays for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12568257/]
- 76. Optimizing viral transduction in immune cell therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12046913/]
- 77. Comparative Analysis of Extracellular Vesicle and Virus Co ... [https://www.mdpi.com/1999-4915/17/11/1470]
- 78. Development and Optimization of an RNA-Isolating ... [https://www.mdpi.com/1422-0067/26/22/11171]
- 79. Cell Density Effects in Different Cell Culture Media and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6631978/]
- 80. Serum-based media concerns in viral vector manufacturing [https://exothera.world/serum-based-media-concerns-in-viral-vector-manufacturing/]
- 81. Adherent Cell Culture Protocol [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-adherent-cells.html]
- 82. Continuous manufacturing of viral particles [https://www.sciencedirect.com/science/article/abs/pii/S2211339818300492]
- 83. Comparison of the efficiency of ultrafiltration, precipitation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10885727/]
- 84. Comparative evaluation of methods for isolating extracellular ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-025-02207-x]
- 85. Cell culture and virus isolation [https://bio-protocol.org/exchange/minidetail?id=7974541&type=30]
- 86. Laboratory Protocol for Propagation and Purification of ... [https://pubmed.ncbi.nlm.nih.gov/40016454/]
- 87. Downstream processing of AAV based gene therapy vectors [https://www.sciencedirect.com/science/article/pii/S138358662501648X]
- 88. Viral viability markers of SARS-CoV-2: a comparison of cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12502786/]
- 89. Virus Detection: A Review of the Current and Emerging ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2021.637559/full]
- 90. Session 4: Infectivity Determination of Viruses: Cell Culture ... [https://watermicro2025.nl/infectivity-determination-of-viruses-cell-culture-vs-molecular-methods-pcr/]
- 91. Contemporary seasonal human coronaviruses display ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12456146/]
- 92. Innovative exploration of Hep-2 cell culture in the isolation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12076762/]
- 93. Monitoring SARS‐CoV‐2 Dissemination in Wastewater and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12401128/]
- 94. Comparison of the methods for isolation and detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10028190/]
- 95. Harmonizing TUNEL with multiplexed iterative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146639/]
- 96. The Validation of a Hemagglutination Inhibition Assay That ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12031337/]
- 97. A Modified Novel Validated High-Throughput ... [https://www.mdpi.com/2076-2607/12/11/2358]
- 98. The Validation of a Hemagglutination Inhibition Assay That ... [https://pubmed.ncbi.nlm.nih.gov/40333216/]
- 99. STRATEGY FOR VALIDATION OF NEW MYCOPLASMA ... [https://pubmed.ncbi.nlm.nih.gov/40962418/]
- 100. Nucleic Acid Testing Validation and Verification [https://clsi.org/resources/insights/nucleic-acid-testing-validation-and-verification/]
- 101. Accessible Color Palette Generator | WCAG Compliant [https://venngage.com/tools/accessible-color-palette-generator]
- 102. An Innovative Binding-Protein-Based dsRNA Extraction ... [https://pubmed.ncbi.nlm.nih.gov/40370068/]
- 103. Optimized Protocol for the Amplification and Viral Titration ... [https://www.protocols.io/view/optimized-protocol-for-the-amplification-and-viral-g999bz997.html]